

Unravelling mechanisms of p53-mediated tumour suppression
- PMID: 24739573
- PMCID: PMC4049238
- DOI: 10.1038/nrc3711
Unravelling mechanisms of p53-mediated tumour suppression
Abstract
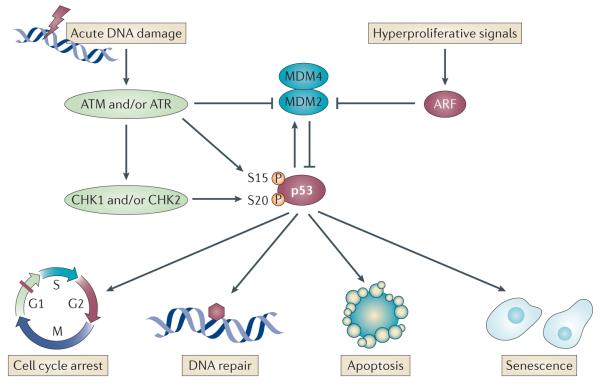
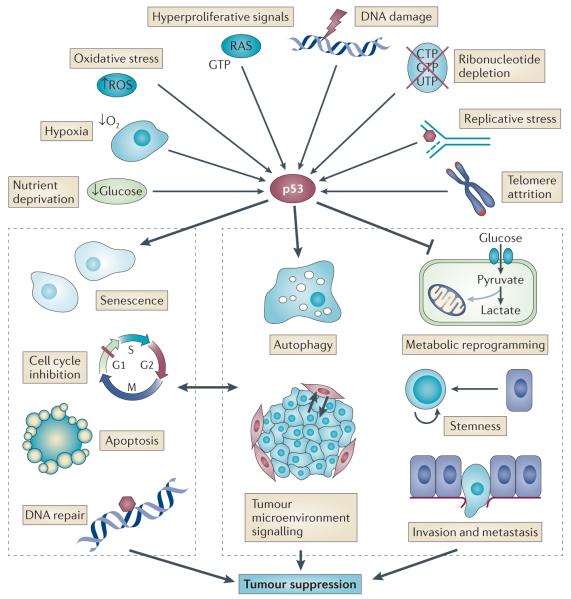
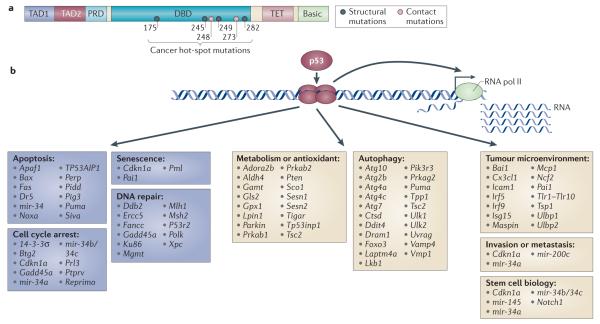
p53 is a crucial tumour suppressor that responds to diverse stress signals by orchestrating specific cellular responses, including transient cell cycle arrest, cellular senescence and apoptosis, which are all processes associated with tumour suppression. However, recent studies have challenged the relative importance of these canonical cellular responses for p53-mediated tumour suppression and have highlighted roles for p53 in modulating other cellular processes, including metabolism, stem cell maintenance, invasion and metastasis, as well as communication within the tumour microenvironment. In this Opinion article, we discuss the roles of classical p53 functions, as well as emerging p53-regulated processes, in tumour suppression.
Figures
References
-
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. - PubMed
-
- Lang GA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. - PubMed
-
- Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature Cell Biol. 2007;9:573–580. - PubMed
-
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
