

From local structure to a global framework: recognition of protein folds
- PMID: 24740960
- PMCID: PMC4006237
- DOI: 10.1098/rsif.2013.1147
From local structure to a global framework: recognition of protein folds
Abstract
Protein folding has been a major area of research for many years. Nonetheless, the mechanisms leading to the formation of an active biological fold are still not fully apprehended. The huge amount of available sequence and structural information provides hints to identify the putative fold for a given sequence. Indeed, protein structures prefer a limited number of local backbone conformations, some being characterized by preferences for certain amino acids. These preferences largely depend on the local structural environment. The prediction of local backbone conformations has become an important factor to correctly identifying the global protein fold. Here, we review the developments in the field of local structure prediction and especially their implication in protein fold recognition.
Keywords: bioinformatics; local structure prediction; protein fold recognition; sequence annotation; structural alphabets; threading.
Figures
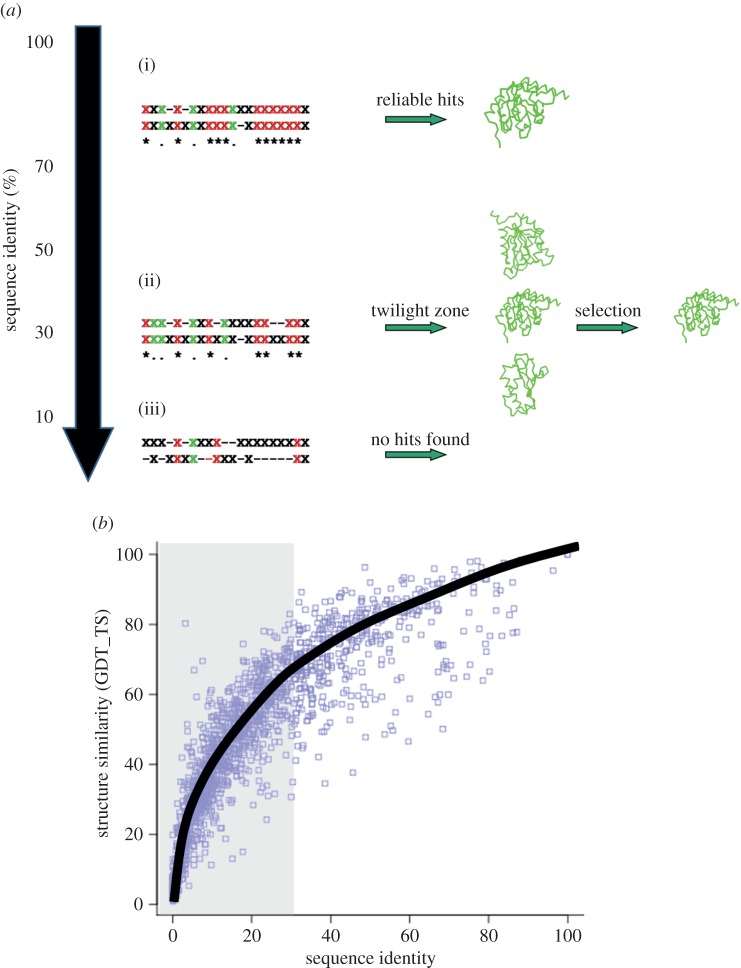
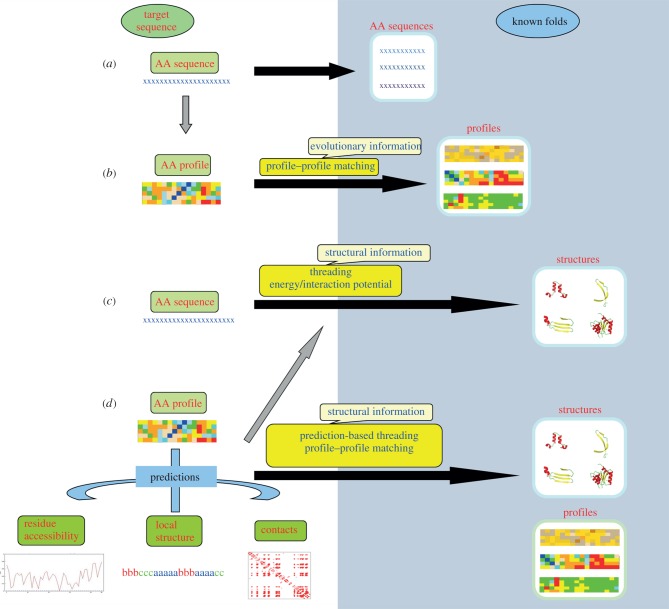
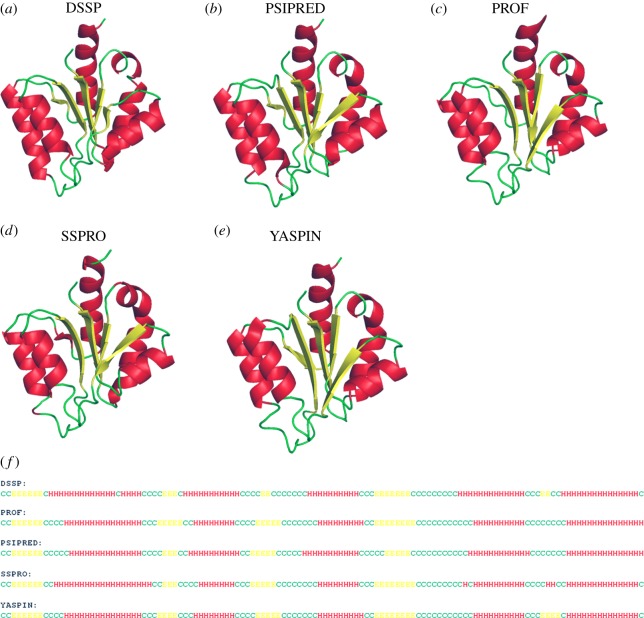
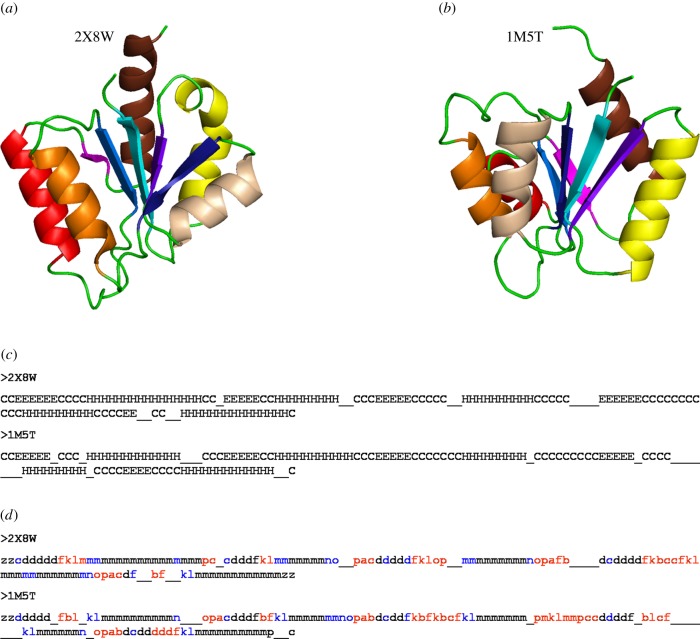
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources