

Inhibition of protein kinase II (CK2) prevents induced signal transducer and activator of transcription (STAT) 1/3 and constitutive STAT3 activation
- PMID: 24742922
- PMCID: PMC4039151
- DOI: 10.18632/oncotarget.1852
Inhibition of protein kinase II (CK2) prevents induced signal transducer and activator of transcription (STAT) 1/3 and constitutive STAT3 activation
Abstract
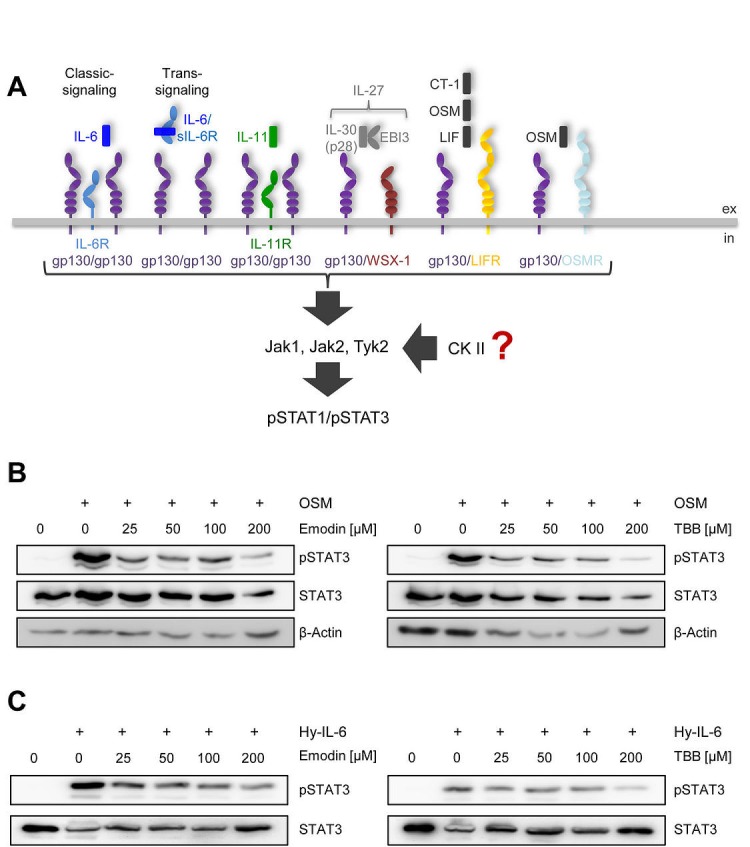
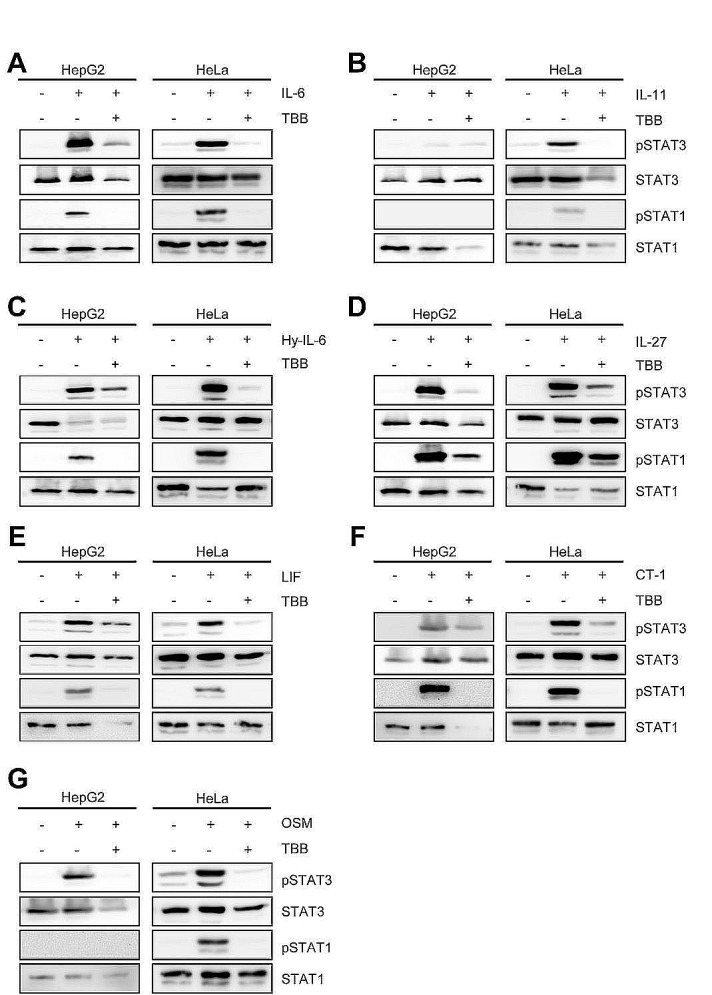
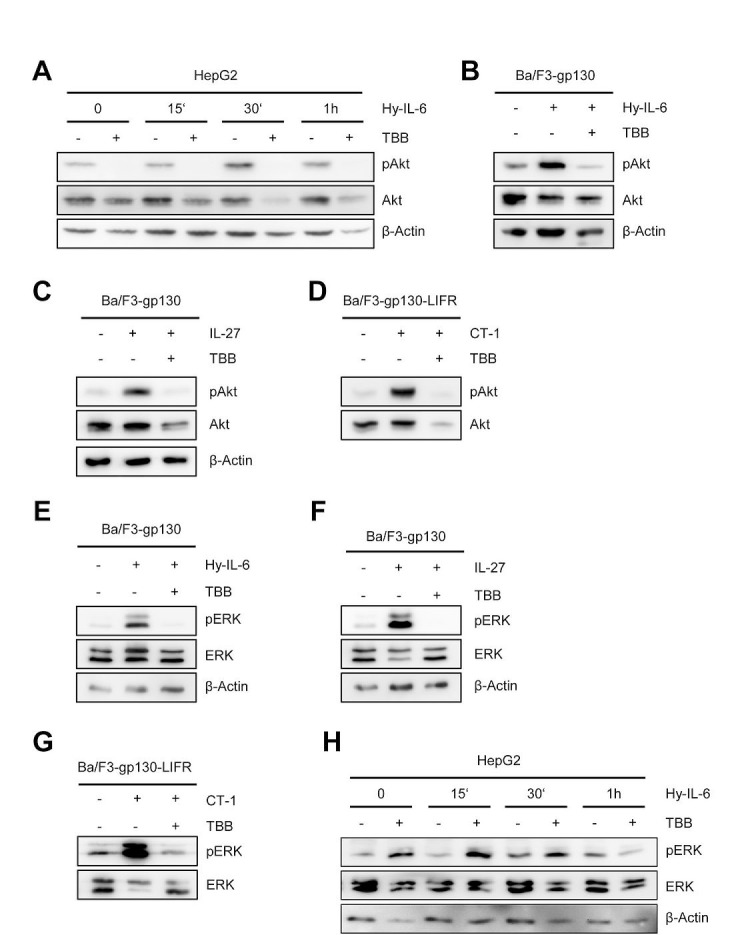
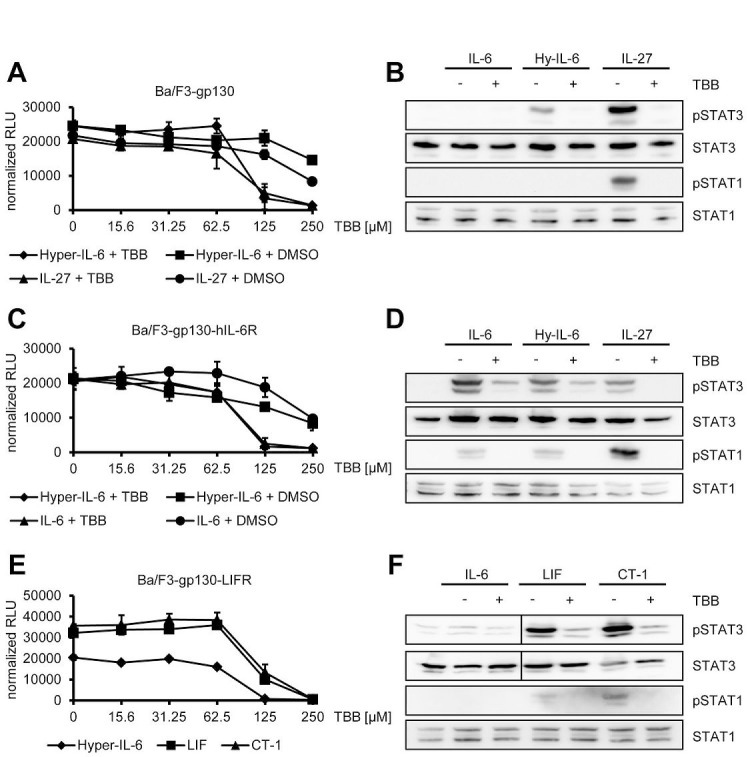
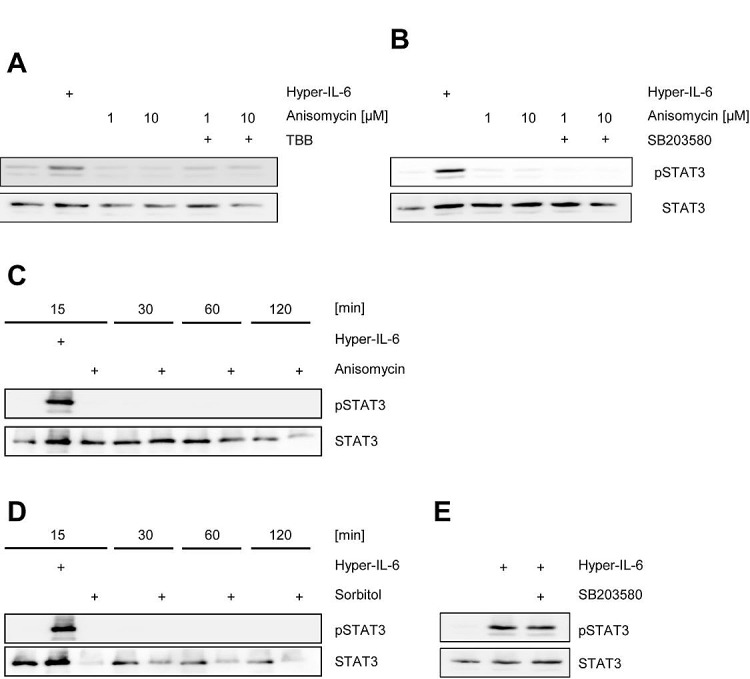
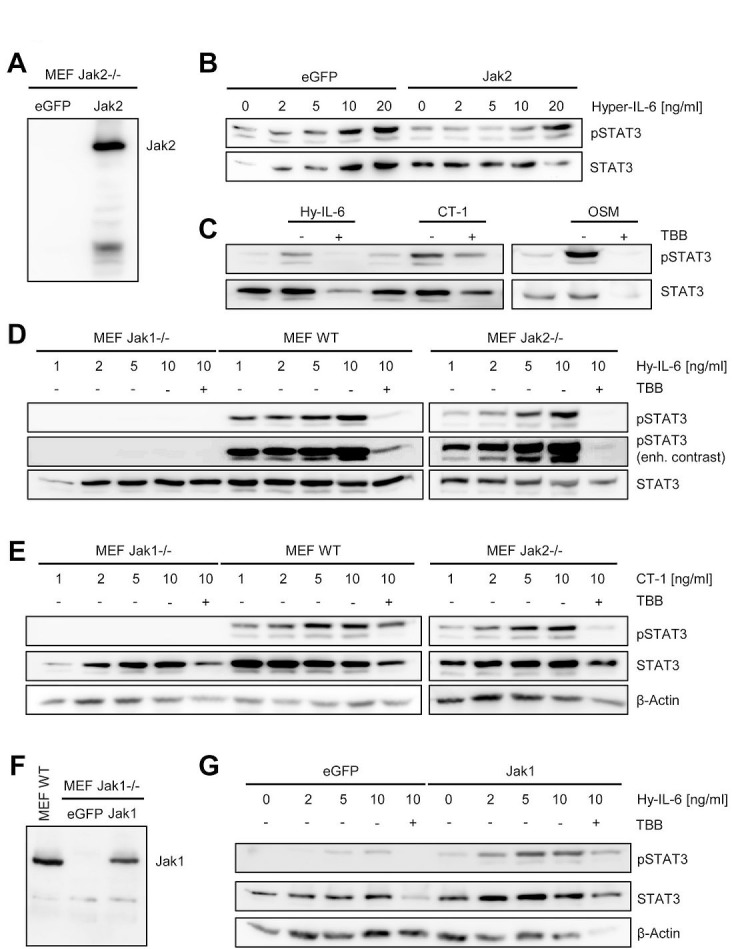
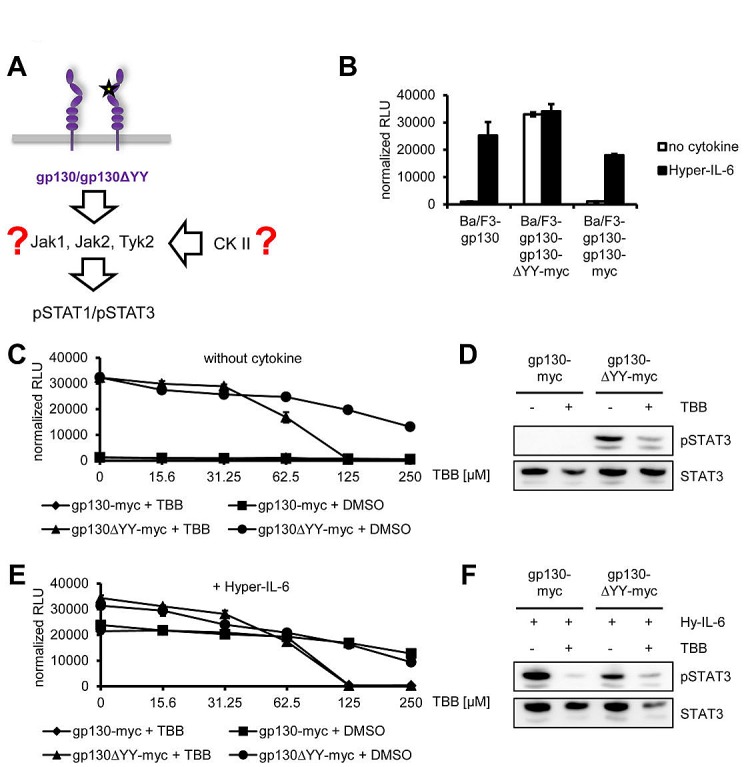
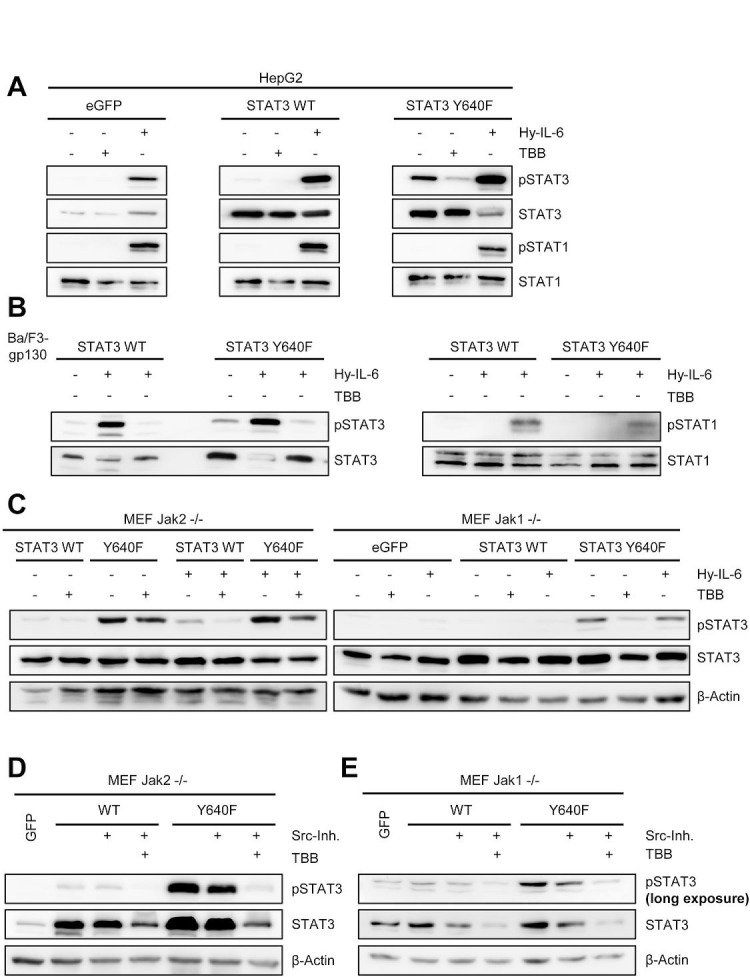
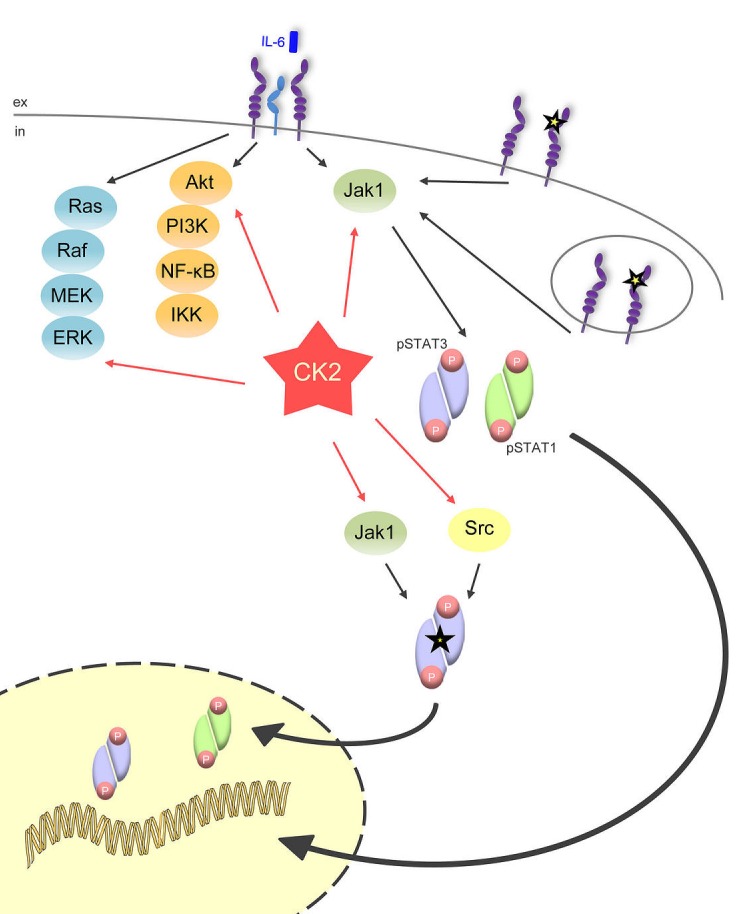
The Janus kinase / signal transducer and activator of transcription (Jak/STAT) pathway can be activated by many different cytokines, among them all members of the Interleukin (IL-)6 family. Dysregulation of this pathway, resulting in its constitutive activation, is associated with chronic inflammation and cancer development. In the present study, we show that activity of protein kinase II (CK2), a ubiquitously expressed serine/threonine kinase, is needed for induced activation of STAT1 and STAT3 by IL-6 classic and trans-signaling, IL-11, IL-27, oncostatin M (OSM), leukemia inhibitory factor (LIF) and cardiotrophin-1 (CT-1). Inhibition of CK2 efficiently prevented STAT phosphorylation and inhibited cytokine-dependent cell proliferation in a Jak1-dependent manner. Conversely, forced activation of CK2 alone was not sufficient to induce activation of the Jak/STAT signaling pathway. Inhibition of CK2 in turn inhibited Jak1-dependent STAT activation by oncogenic gp130 mutations. Furthermore, CK2 inhibition diminished the Jak1- and Src kinase-dependent phosphorylation of a constitutively active STAT3 mutant recently described in human large granular lymphocytic leukemia. In conclusion, we characterize CK2 as an essential component of the Jak/STAT pathway. Pharmacologic inhibition of this kinase is therefore a promising strategy to treat human inflammatory diseases and malignancies associated with constitutive activation of the Jak/STAT pathway.
Figures
References
-
- Garbers C, Hermanns H, Schaper F, Müller-Newen G, Grötzinger J, Rose-John S, Scheller J. Plasticity and cross-talk of Interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012;23(3):85–182. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
