

Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation
- PMID: 24743168
- PMCID: PMC3990503
- DOI: 10.1371/journal.pgen.1004261
Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation
Abstract
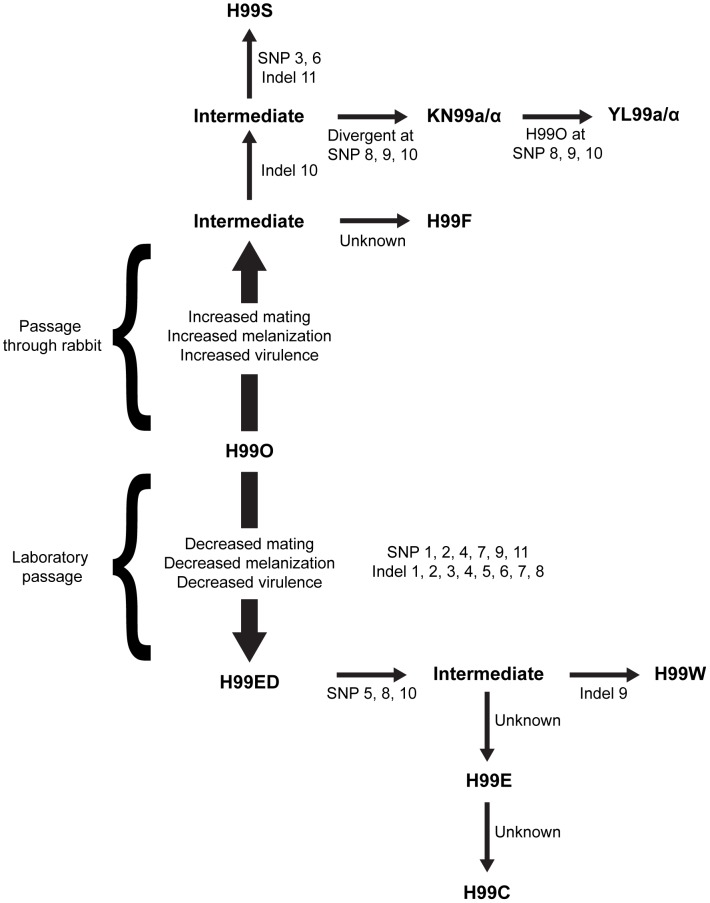
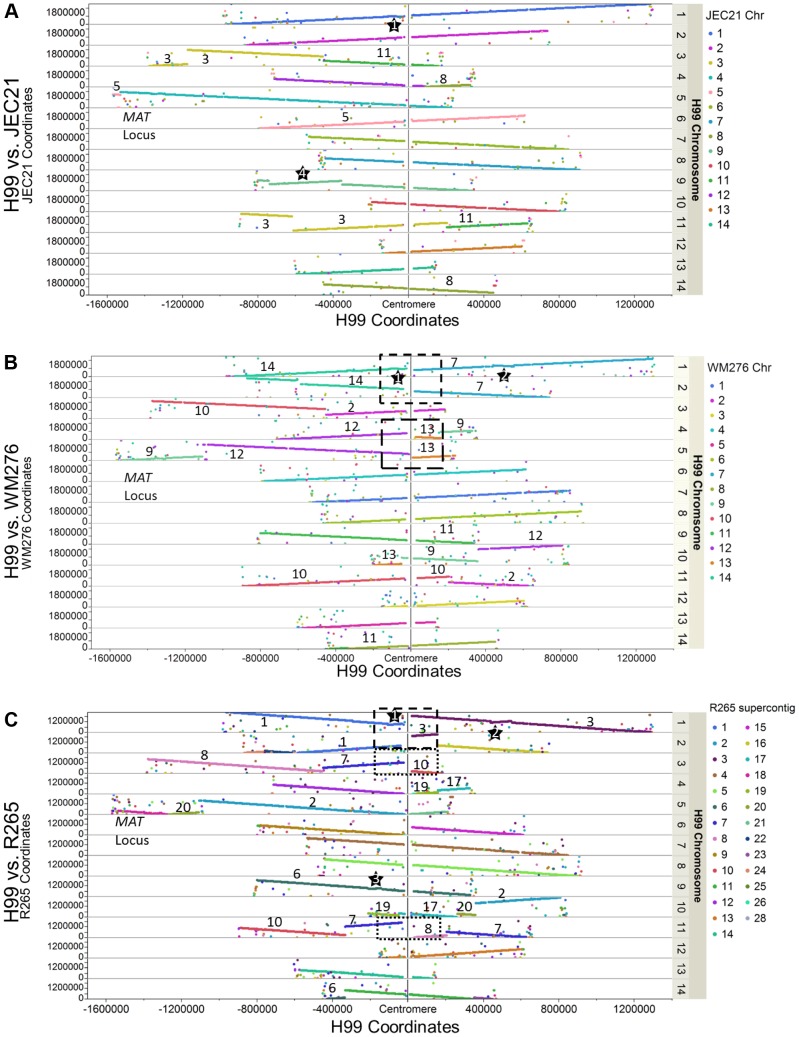
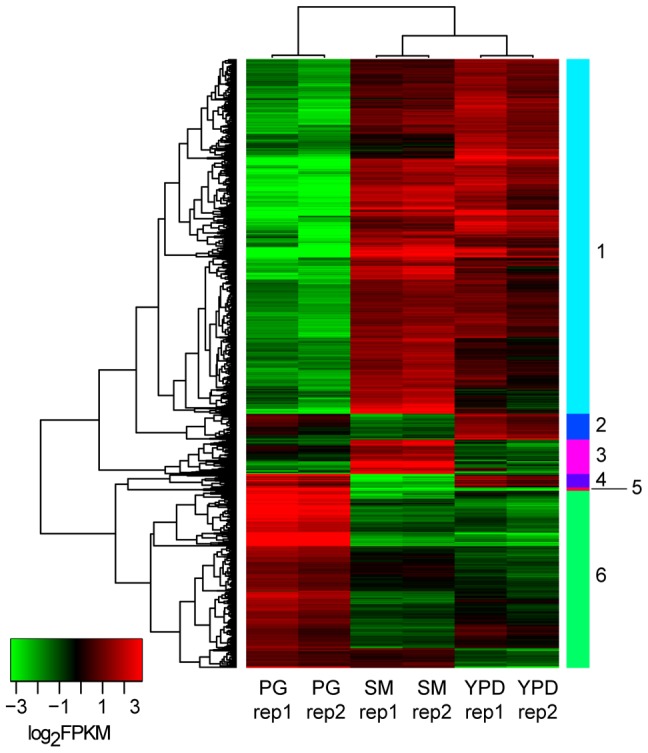
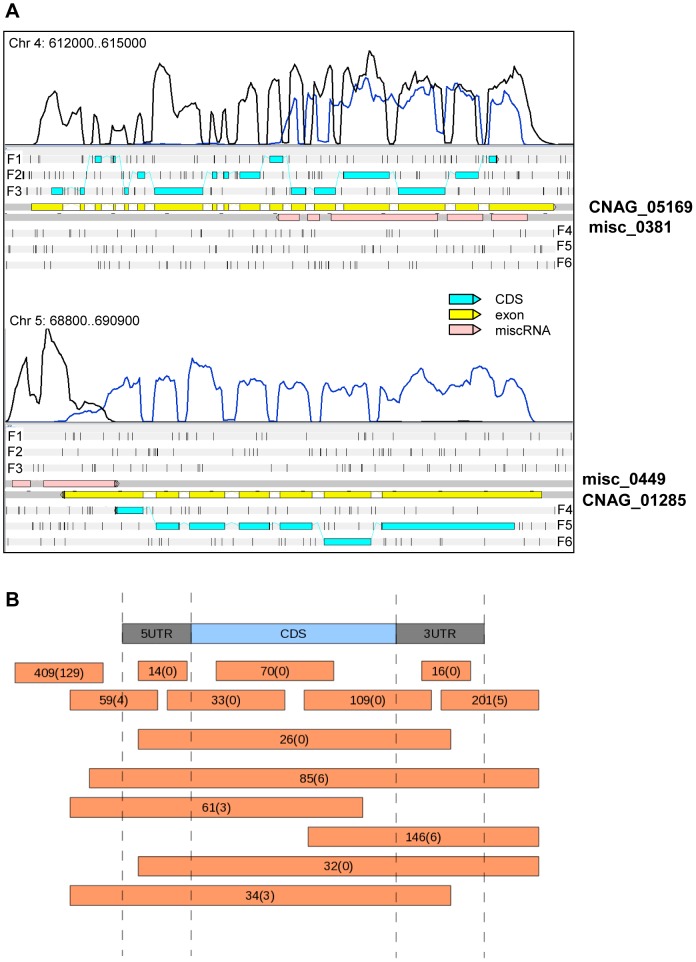
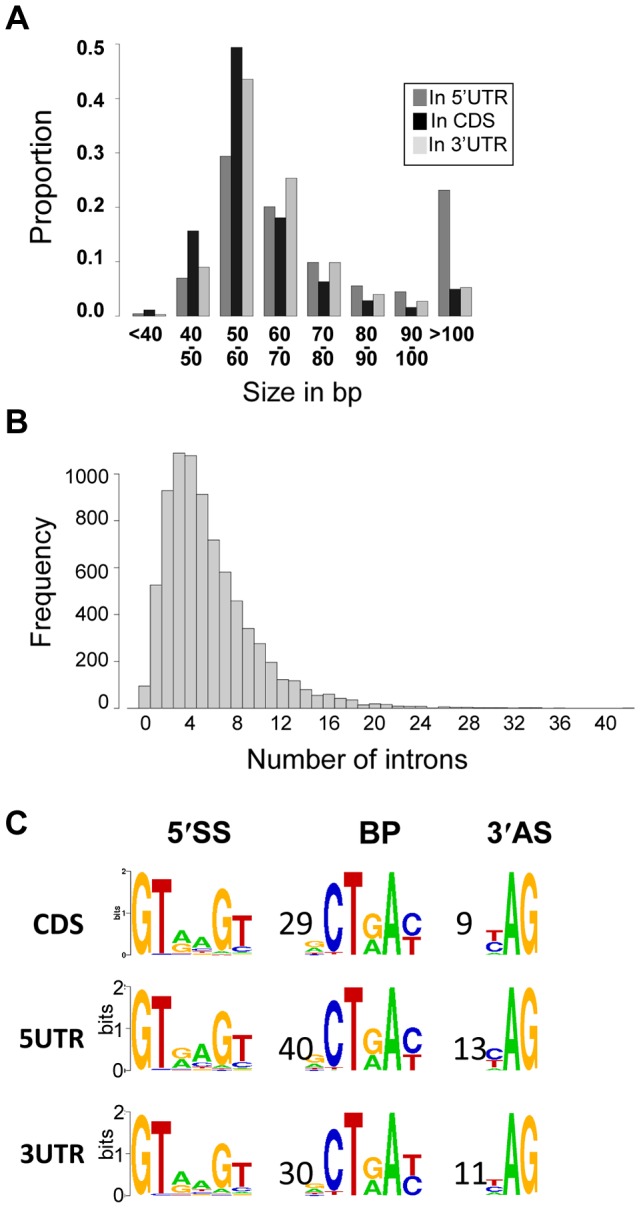
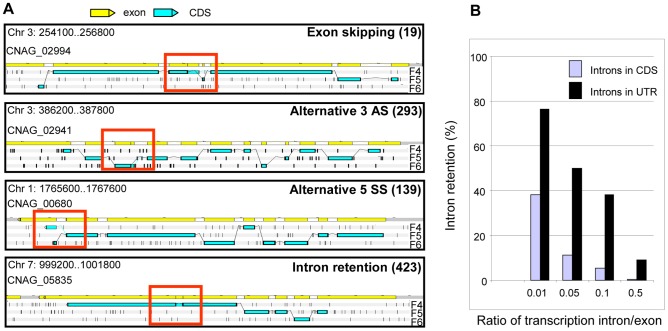
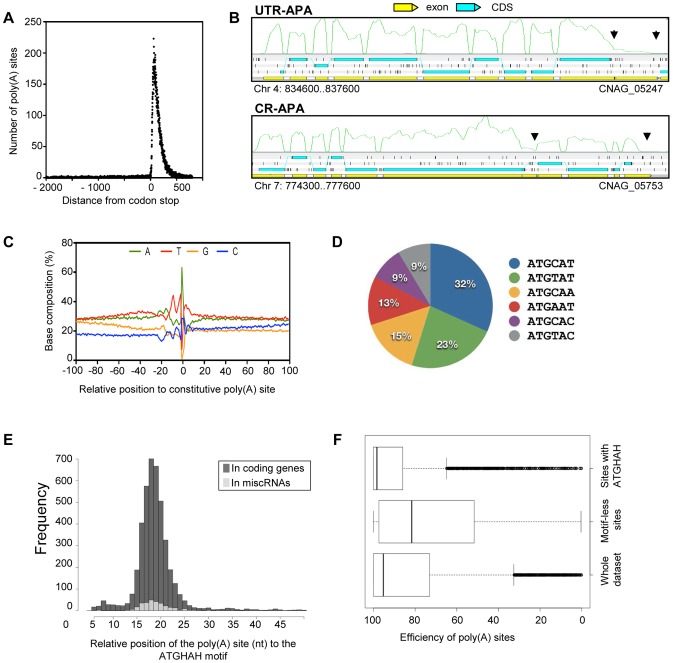
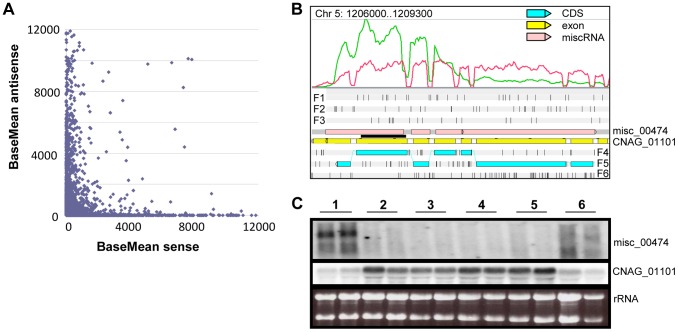
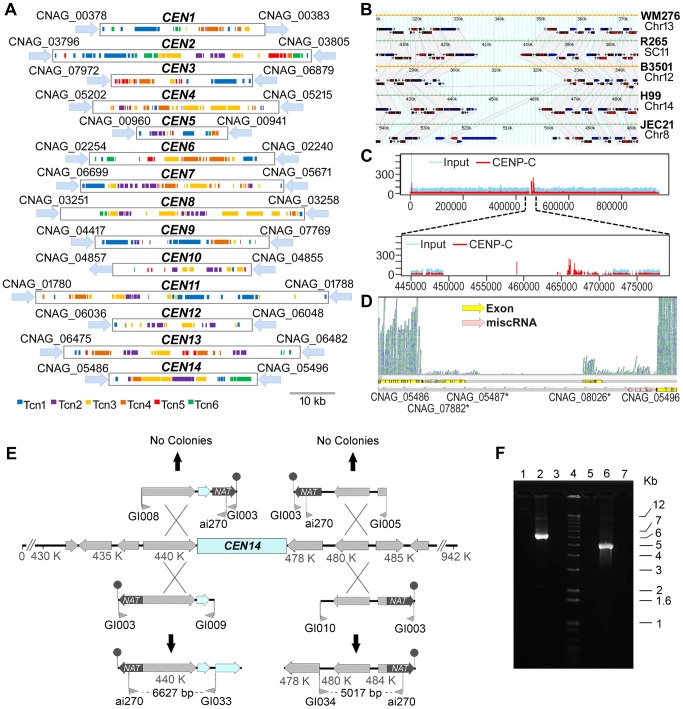
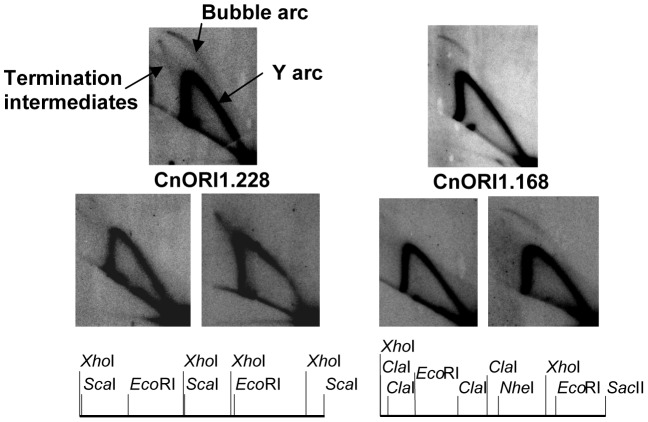
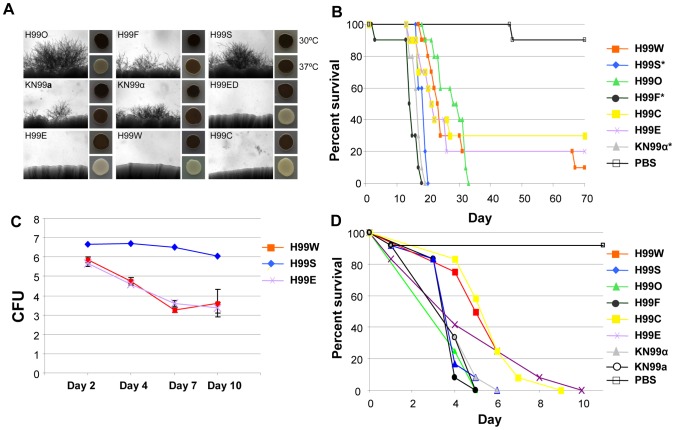
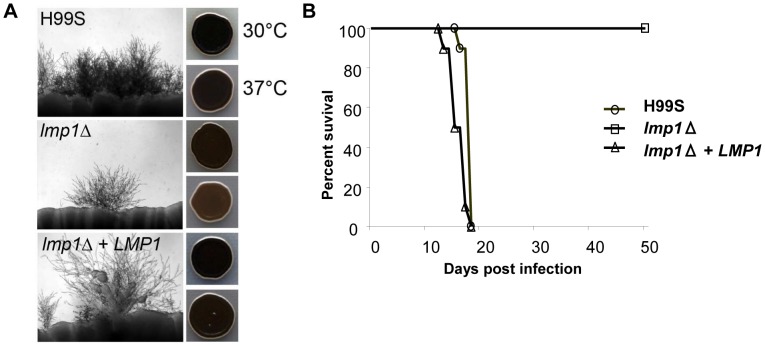
Cryptococcus neoformans is a pathogenic basidiomycetous yeast responsible for more than 600,000 deaths each year. It occurs as two serotypes (A and D) representing two varieties (i.e. grubii and neoformans, respectively). Here, we sequenced the genome and performed an RNA-Seq-based analysis of the C. neoformans var. grubii transcriptome structure. We determined the chromosomal locations, analyzed the sequence/structural features of the centromeres, and identified origins of replication. The genome was annotated based on automated and manual curation. More than 40,000 introns populating more than 99% of the expressed genes were identified. Although most of these introns are located in the coding DNA sequences (CDS), over 2,000 introns in the untranslated regions (UTRs) were also identified. Poly(A)-containing reads were employed to locate the polyadenylation sites of more than 80% of the genes. Examination of the sequences around these sites revealed a new poly(A)-site-associated motif (AUGHAH). In addition, 1,197 miscRNAs were identified. These miscRNAs can be spliced and/or polyadenylated, but do not appear to have obvious coding capacities. Finally, this genome sequence enabled a comparative analysis of strain H99 variants obtained after laboratory passage. The spectrum of mutations identified provides insights into the genetics underlying the micro-evolution of a laboratory strain, and identifies mutations involved in stress responses, mating efficiency, and virulence.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Brown GD, Denning DW, Levitz SM (2012) Tackling human fungal infections. Science 336: 647. - PubMed
-
- Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, et al. (2009) Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23: 525–530. - PubMed
-
- Kwon-Chung KJ, Boekhout T, Wickes BL, Fell JW, Systematics of the genus Cryptococcus and its type species C. neoformans, in Cryptococcus from human pathogen to model yeast, J. Heitman, et al., Editors. 2011, ASM Press: Washington DC. p.3–15.
-
- Heitman J, Casadevall A, Lodge JK, Perfect JR (1999) The Cryptococcus neoformans genome sequencing project. Mycopathologia 148: 1–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- T32 GM007184/GM/NIGMS NIH HHS/United States
- R01 AI039115/AI/NIAID NIH HHS/United States
- AI50113-10/AI/NIAID NIH HHS/United States
- U54HG003067/HG/NHGRI NIH HHS/United States
- R01 AI050184/AI/NIAID NIH HHS/United States
- R21 AI094364/AI/NIAID NIH HHS/United States
- R01 AI050113/AI/NIAID NIH HHS/United States
- R01 NS042263/NS/NINDS NIH HHS/United States
- AI094364/AI/NIAID NIH HHS/United States
- NS042263/NS/NINDS NIH HHS/United States
- R37 AI039115/AI/NIAID NIH HHS/United States
- R01 AI073896/AI/NIAID NIH HHS/United States
- R01 AI093257/AI/NIAID NIH HHS/United States
- HHSN272200900018C/AI/NIAID NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- T32 GM007754/GM/NIGMS NIH HHS/United States
- AI39115-16/AI/NIAID NIH HHS/United States
- R01 AI053721/AI/NIAID NIH HHS/United States
- T32 AI052080/AI/NIAID NIH HHS/United States
- R01 AI080275/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
