

Phylogenetic quantification of intra-tumour heterogeneity
- PMID: 24743184
- PMCID: PMC3990475
- DOI: 10.1371/journal.pcbi.1003535
Phylogenetic quantification of intra-tumour heterogeneity
Abstract
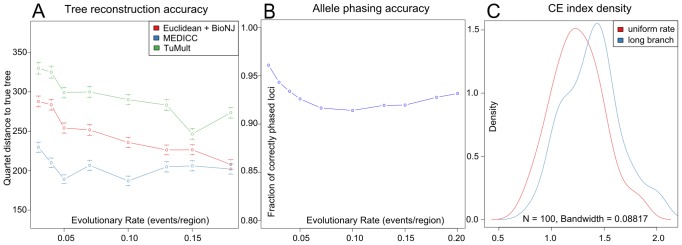
Intra-tumour genetic heterogeneity is the result of ongoing evolutionary change within each cancer. The expansion of genetically distinct sub-clonal populations may explain the emergence of drug resistance, and if so, would have prognostic and predictive utility. However, methods for objectively quantifying tumour heterogeneity have been missing and are particularly difficult to establish in cancers where predominant copy number variation prevents accurate phylogenetic reconstruction owing to horizontal dependencies caused by long and cascading genomic rearrangements. To address these challenges, we present MEDICC, a method for phylogenetic reconstruction and heterogeneity quantification based on a Minimum Event Distance for Intra-tumour Copy-number Comparisons. Using a transducer-based pairwise comparison function, we determine optimal phasing of major and minor alleles, as well as evolutionary distances between samples, and are able to reconstruct ancestral genomes. Rigorous simulations and an extensive clinical study show the power of our method, which outperforms state-of-the-art competitors in reconstruction accuracy, and additionally allows unbiased numerical quantification of tumour heterogeneity. Accurate quantification and evolutionary inference are essential to understand the functional consequences of tumour heterogeneity. The MEDICC algorithms are independent of the experimental techniques used and are applicable to both next-generation sequencing and array CGH data.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
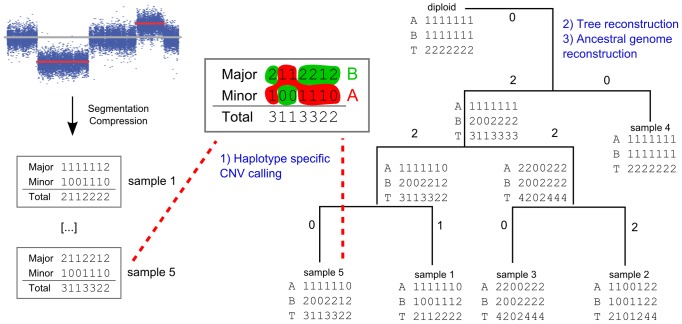
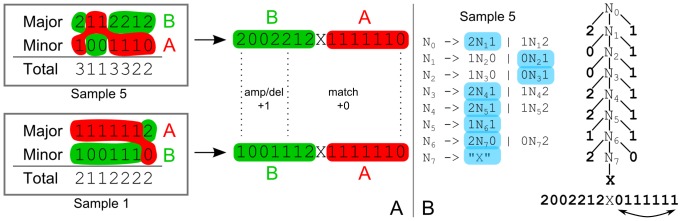
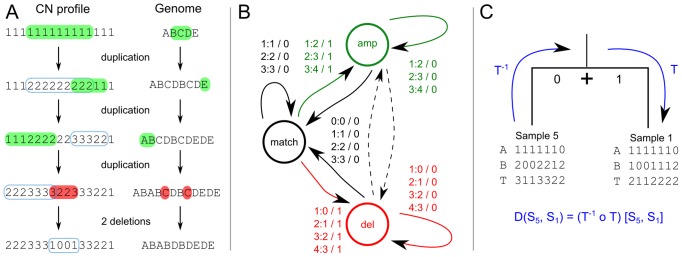
 times with itself to create the the full minimum event FST
times with itself to create the the full minimum event FST  . Edge labels consist of an input symbol, a colon and the corresponding output symbol, followed by a slash and the weight associated with taking that transition. C) The minimum event FST
. Edge labels consist of an input symbol, a colon and the corresponding output symbol, followed by a slash and the weight associated with taking that transition. C) The minimum event FST  is asymmetric and describes the evolution of a genomic profile from its ancestor. Composed with its inverse this yields the symmetric minimum event distance
is asymmetric and describes the evolution of a genomic profile from its ancestor. Composed with its inverse this yields the symmetric minimum event distance  .
.
References
-
- Khalique L, Ayhan A, Weale ME, Jacobs IJ, Ramus SJ, et al. (2007) Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J Pathol 211: 286–295. - PubMed
-
- Khalique L, Ayhan A, Whittaker JC, Singh N, Jacobs IJ, et al. (2009) The clonal evolution of metastases from primary serous epithelial ovarian cancers. Int J Cancer 124: 1579–1586. - PubMed
-
- Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, et al. (2009) Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461: 809–813. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
