

Functional deregulation of KIT: link to mast cell proliferative diseases and other neoplasms
- PMID: 24745671
- PMCID: PMC3994404
- DOI: 10.1016/j.iac.2014.01.002
Functional deregulation of KIT: link to mast cell proliferative diseases and other neoplasms
Abstract
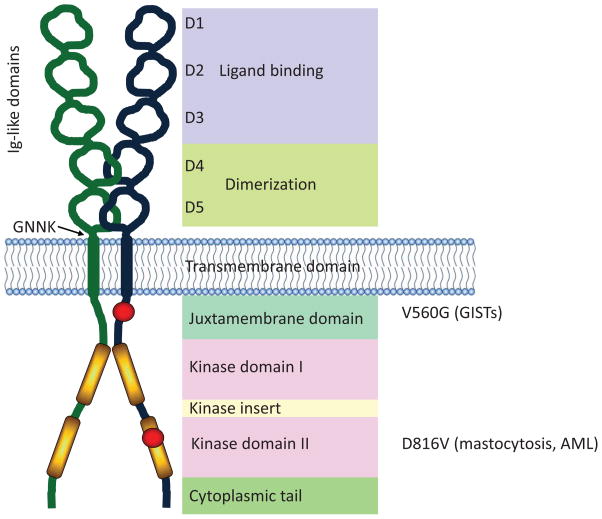
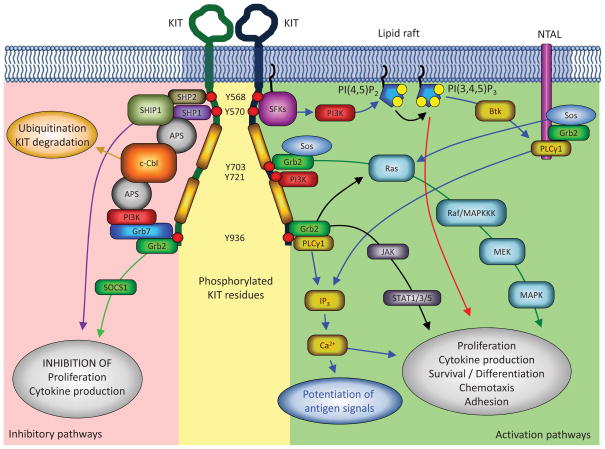
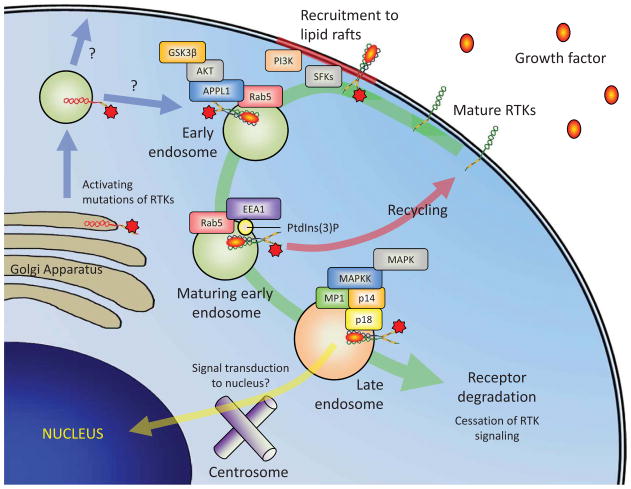
In this review, the authors discuss common gain-of-function mutations in the stem cell factor receptor KIT found in mast cell proliferation disorders and summarize the current understanding of the molecular mechanisms by which these transforming mutations may affect KIT structure and function leading to altered downstream signaling and cellular transformation. Drugs targeting KIT have shown mixed success in the treatment of mastocytosis and other hyperproliferative diseases. A brief overview of the most common KIT inhibitors currently used, the reasons for the varied clinical results of such inhibitors and a discussion of potential new strategies are provided.
Keywords: KIT inhibitors; KIT mutations; KIT signaling; KIT trafficking; Mastocytosis.
Published by Elsevier Inc.
Figures
References
-
- Flanagan JG, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63:185–194. - PubMed
-
- Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. - PubMed
-
- Toksoz D, Zsebo KM, Smith KA, et al. Support of human hematopoiesis in long-term bone marrow cultures by murine stromal cells selectively expressing the membrane-bound and secreted forms of the human homolog of the steel gene product, stem cell factor. Proc Natl Acad Sci U S A. 1992;89:7350–7354. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
