

Self-eating in the plaque: what macrophage autophagy reveals about atherosclerosis
- PMID: 24746519
- PMCID: PMC4061377
- DOI: 10.1016/j.tem.2014.03.010
Self-eating in the plaque: what macrophage autophagy reveals about atherosclerosis
Abstract
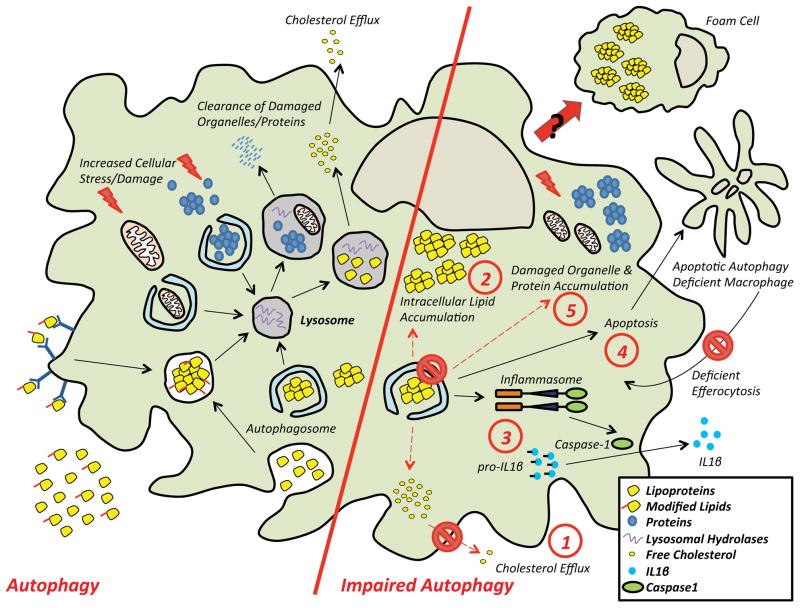
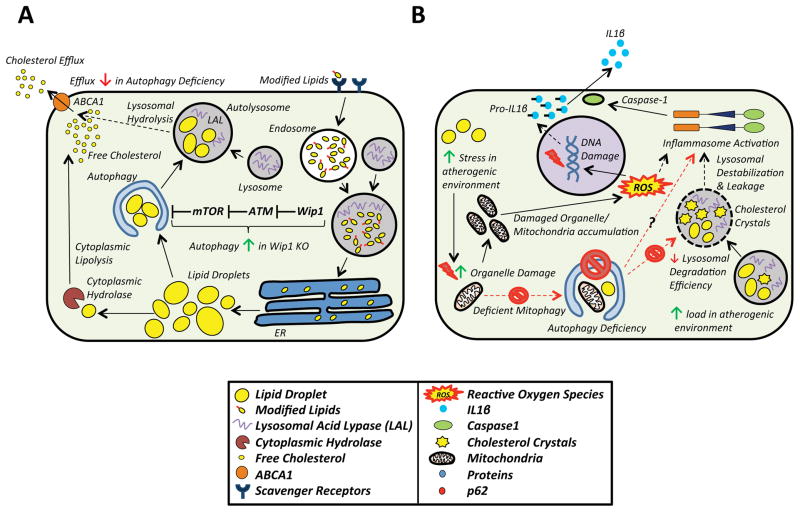
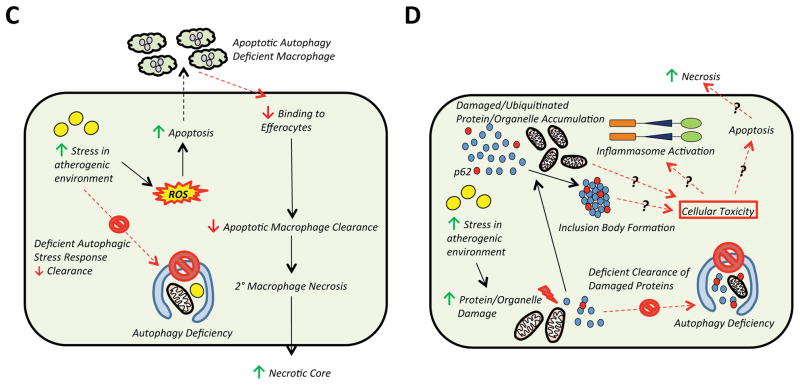
Autophagy (or 'self-eating') is the process by which cellular contents are recycled to support downstream metabolism. An explosion in research in the past decade has implicated its role in both health and disease and established the importance of the autophagic response during periods of stress and nutrient deprivation. Atherosclerosis is a state where chronic exposure to cellular stressors promotes disease progression, and alterations in autophagy are predicted to be consequential. Recent reports linking macrophage autophagy to lipid metabolism, blunted inflammatory signaling, and an overall suppression of proatherogenic processes support this notion. We review these data and provide a framework for understanding the role of macrophage autophagy in the pathogenesis of atherosclerosis, one of the most formidable diseases of our time.
Keywords: atherosclerosis; autophagy; inflammation; lipid metabolism; macrophage.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures


References
-
- Go AS, et al. Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127(1):143–52. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
