

Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells
- PMID: 24748663
- PMCID: PMC4066747
- DOI: 10.1093/nar/gku289
Efficient chromosomal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells
Abstract
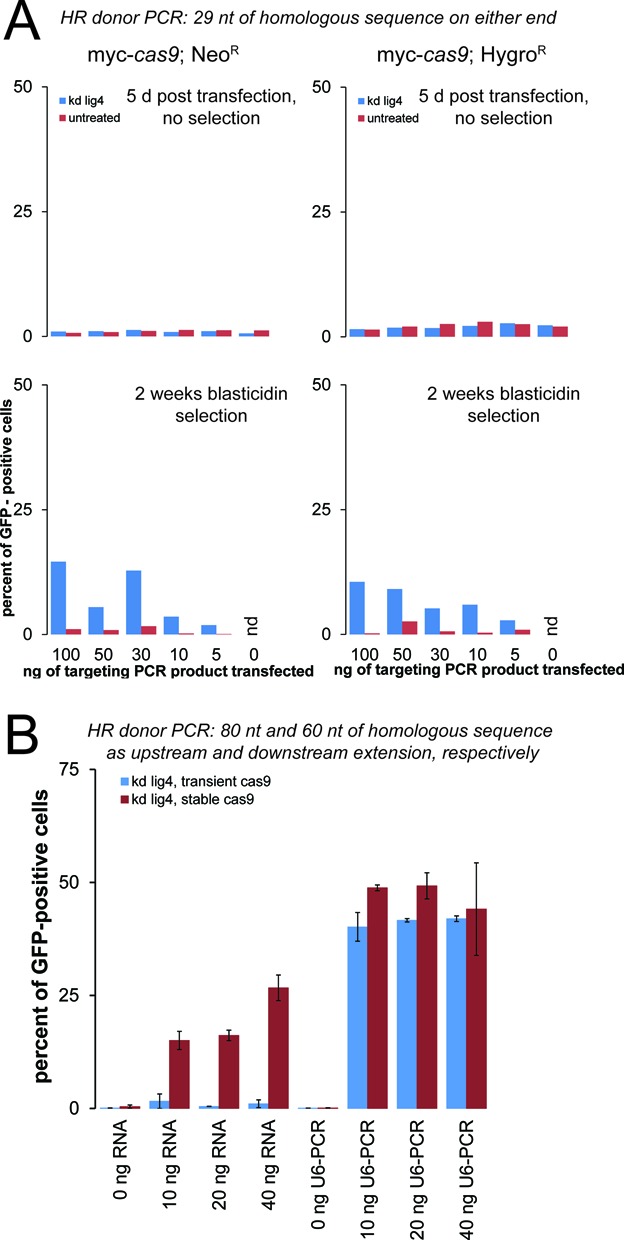
The ability to edit the genome is essential for many state-of-the-art experimental paradigms. Since DNA breaks stimulate repair, they can be exploited to target site-specific integration. The clustered, regularly interspaced, short palindromic repeats (CRISPR)/cas9 system from Streptococcus pyogenes has been harnessed into an efficient and programmable nuclease for eukaryotic cells. We thus combined DNA cleavage by cas9, the generation of homologous recombination donors by polymerase chain reaction (PCR) and transient depletion of the non-homologous end joining factor lig4. Using cultured Drosophila melanogaster S2-cells and the phosphoglycerate kinase gene as a model, we reached targeted integration frequencies of up to 50% in drug-selected cell populations. Homology arms as short as 29 nt appended to the PCR primer resulted in detectable integration, slightly longer extensions are beneficial. We confirmed established rules for S. pyogenes cas9 sgRNA design and demonstrate that the complementarity region allows length variation and 5'-extensions. This enables generation of U6-promoter fusion templates by overlap-extension PCR with a standardized protocol. We present a series of PCR template vectors for C-terminal protein tagging and clonal Drosophila S2 cell lines with stable expression of a myc-tagged cas9 protein. The system can be used for epitope tagging or reporter gene knock-ins in an experimental setup that can in principle be fully automated.
© The Author(s) 2014. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. - PubMed
-
- Garneau J.E., Dupuis M.E., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadan A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
