

Efficient Bayesian inference under the structured coalescent
- PMID: 24753484
- PMCID: PMC4207426
- DOI: 10.1093/bioinformatics/btu201
Efficient Bayesian inference under the structured coalescent
Abstract
Motivation: Population structure significantly affects evolutionary dynamics. Such structure may be due to spatial segregation, but may also reflect any other gene-flow-limiting aspect of a model. In combination with the structured coalescent, this fact can be used to inform phylogenetic tree reconstruction, as well as to infer parameters such as migration rates and subpopulation sizes from annotated sequence data. However, conducting Bayesian inference under the structured coalescent is impeded by the difficulty of constructing Markov Chain Monte Carlo (MCMC) sampling algorithms (samplers) capable of efficiently exploring the state space.
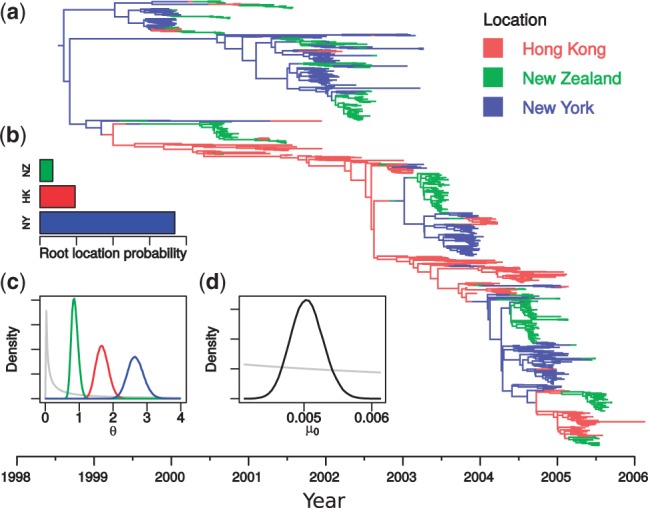
Results: In this article, we present a new MCMC sampler capable of sampling from posterior distributions over structured trees: timed phylogenetic trees in which lineages are associated with the distinct subpopulation in which they lie. The sampler includes a set of MCMC proposal functions that offer significant mixing improvements over a previously published method. Furthermore, its implementation as a BEAST 2 package ensures maximum flexibility with respect to model and prior specification. We demonstrate the usefulness of this new sampler by using it to infer migration rates and effective population sizes of H3N2 influenza between New Zealand, New York and Hong Kong from publicly available hemagglutinin (HA) gene sequences under the structured coalescent.
Availability and implementation: The sampler has been implemented as a publicly available BEAST 2 package that is distributed under version 3 of the GNU General Public License at http://compevol.github.io/MultiTypeTree.
© The Author 2014. Published by Oxford University Press.
Figures
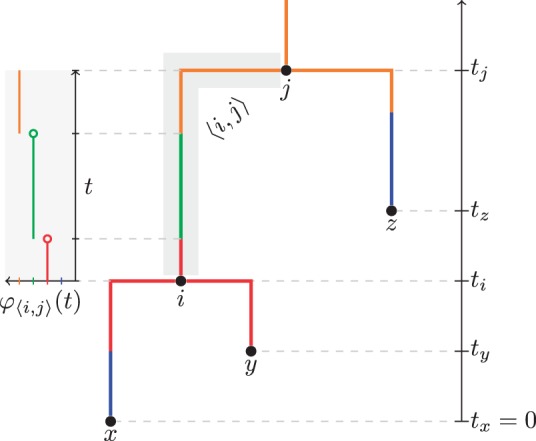
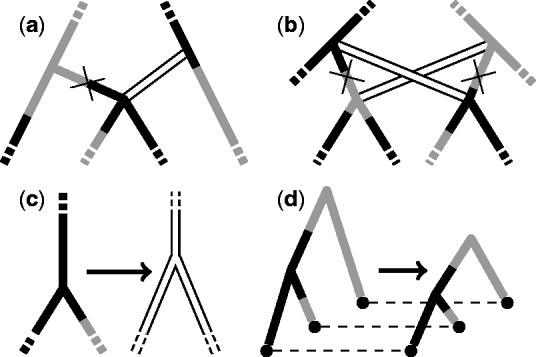
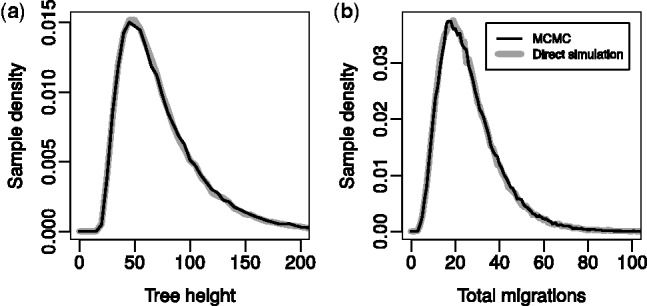
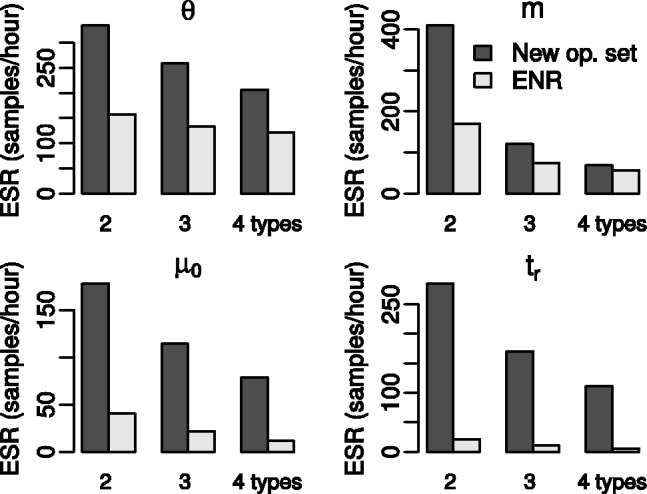
References
-
- Baum LE, et al. A maximization technique occurring in the statistical analysis of probabilistic functions of Markov chains. Ann. Math. Stat. 1970;41:164.
-
- Beerli P. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics. 2006;22:341–345. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
