

Analytical validation of whole exome and whole genome sequencing for clinical applications
- PMID: 24758382
- PMCID: PMC4022392
- DOI: 10.1186/1755-8794-7-20
Analytical validation of whole exome and whole genome sequencing for clinical applications
Abstract
Background: Whole exome and genome sequencing (WES/WGS) is now routinely offered as a clinical test by a growing number of laboratories. As part of the test design process each laboratory must determine the performance characteristics of the platform, test and informatics pipeline. This report documents one such characterization of WES/WGS.
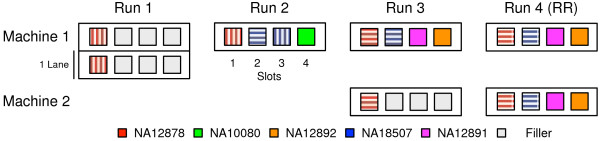
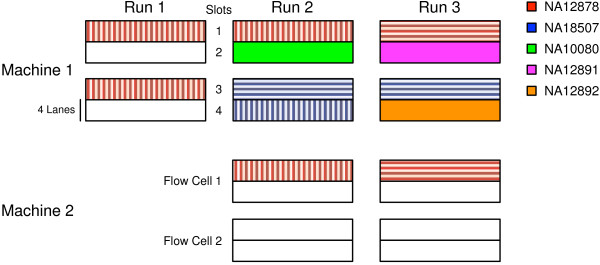
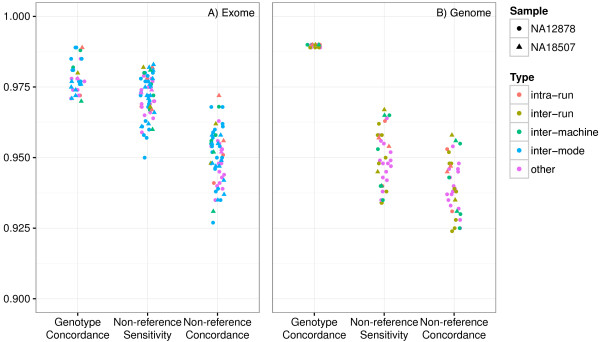
Methods: Whole exome and whole genome sequencing was performed on multiple technical replicates of five reference samples using the Illumina HiSeq 2000/2500. The sequencing data was processed with a GATK-based genome analysis pipeline to evaluate: intra-run, inter-run, inter-mode, inter-machine and inter-library consistency, concordance with orthogonal technologies (microarray, Sanger) and sensitivity and accuracy relative to known variant sets.
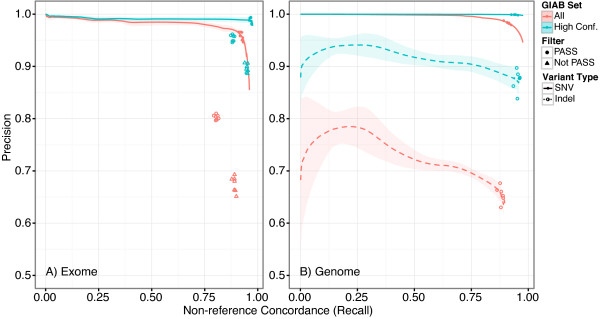
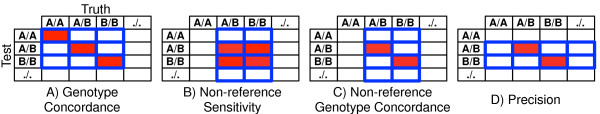
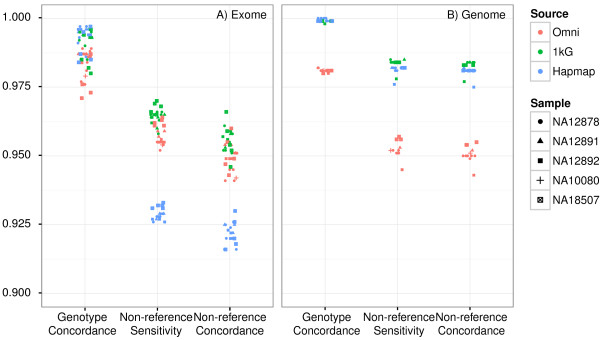
Results: Concordance to high-density microarrays consistently exceeds 97% (and typically exceeds 99%) and concordance between sequencing replicates also exceeds 97%, with no observable differences between different flow cells, runs, machines or modes. Sensitivity relative to high-density microarray variants exceeds 95%. In a detailed study of a 129 kb region, sensitivity was lower with some validated single-base insertions and deletions "not called". Different variants are "not called" in each replicate: of all variants identified in WES data from the NA12878 reference sample 74% of indels and 89% of SNVs were called in all seven replicates, in NA12878 WGS 52% of indels and 88% of SNVs were called in all six replicates. Key sources of non-uniformity are variance in depth of coverage, artifactual variants resulting from repetitive regions and larger structural variants.
Conclusion: We report a comprehensive performance characterization of WES/WGS that will be relevant to offering laboratories, consumers of genome sequencing and others interested in the analytical validity of this technology.
Figures
References
-
- Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, Lu F, Lyon E, Voelkerding KV, Zehnbauer BA, Agarwala R, Bennett SF, Chen B, Chin ELH, Compton JG, Das S, Farkas DH, Ferber MJ, Funke BH, Furtado MR, Ganova-Raeva LM, Geigenmüller U, Gunselman SJ, Hegde MR, Johnson PLF, Kasarskis A, Kulkarni S, Lenk T, Liu CSJ, Manion M. et al.Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012;7:1033–1036. doi: 10.1038/nbt.2403. - DOI - PMC - PubMed
-
- Rieber N, Zapatka M, Lasitschka B, Jones D, Northcott P, Hutter B, Jäger N, Kool M, Taylor M, Lichter P, Pfister S, Wolf S, Brors B, Eils R. Coverage bias and sensitivity of variant calling for four whole-genome sequencing technologies. PLoS One. 2013;7:e66621. doi: 10.1371/journal.pone.0066621. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
