

The instability of the BTB-KELCH protein Gigaxonin causes Giant Axonal Neuropathy and constitutes a new penetrant and specific diagnostic test
- PMID: 24758703
- PMCID: PMC4234992
- DOI: 10.1186/2051-5960-2-47
The instability of the BTB-KELCH protein Gigaxonin causes Giant Axonal Neuropathy and constitutes a new penetrant and specific diagnostic test
Abstract
Background: The BTB-KELCH protein Gigaxonin plays key roles in sustaining neuron survival and cytoskeleton architecture. Indeed, recessive mutations in the Gigaxonin-encoding gene cause Giant Axonal Neuropathy (GAN), a severe neurodegenerative disorder characterized by a wide disorganization of the Intermediate Filament network. Growing evidences suggest that GAN is a continuum with the peripheral neuropathy Charcot-Marie-Tooth diseases type 2 (CMT2). Sharing similar sensory-motor alterations and aggregation of Neurofilaments, few reports have revealed that GAN and some CMT2 forms can be misdiagnosed on clinical and histopathological examination. The goal of this study is to propose a new differential diagnostic test for GAN/CMT2. Moreover, we aim at identifying the mechanisms causing the loss-of-function of Gigaxonin, which has been proposed to bind CUL3 and substrates as part of an E3 ligase complex.
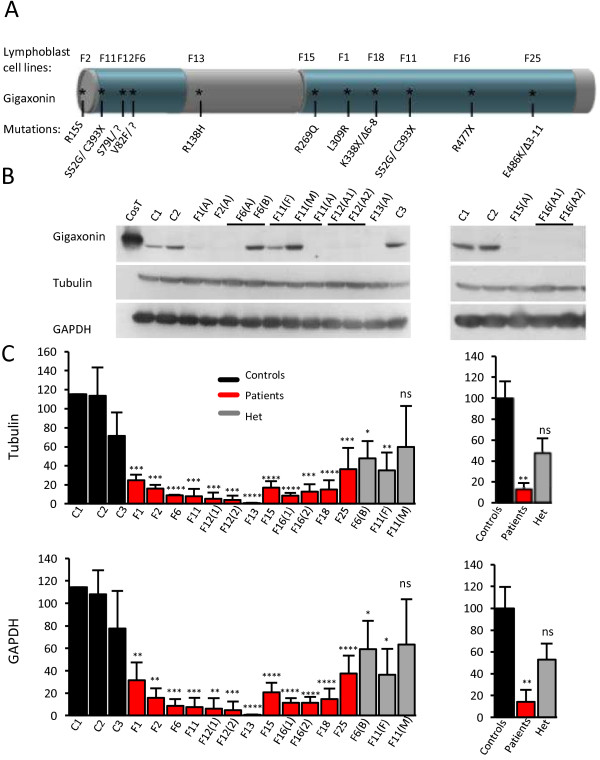
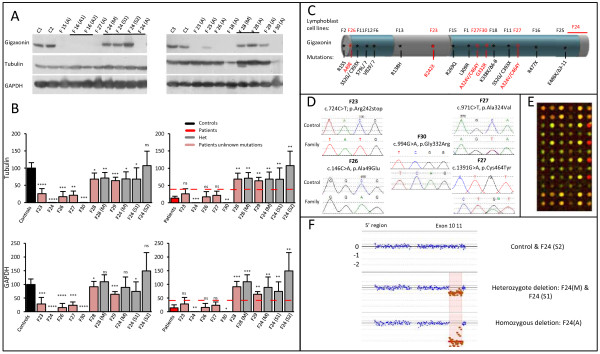
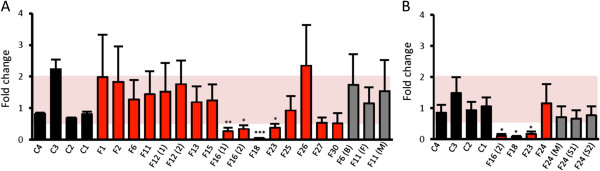
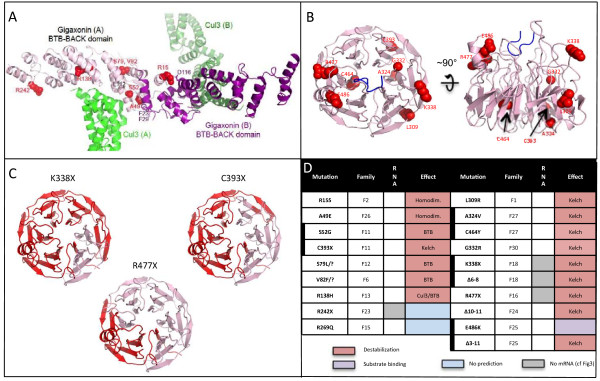
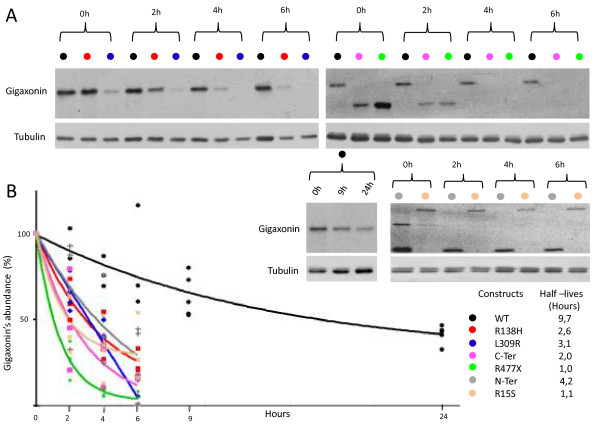
Results: We establish that determining Gigaxonin level constitutes a very valuable diagnostic test in discriminating new GAN cases from clinically related inherited neuropathies. Indeed, in a set of seven new families presenting a neuropathy resembling GAN/CMT2, only five exhibiting a reduced Gigaxonin abundance have been subsequently genetically linked to GAN. Generating the homology modeling of Gigaxonin, we suggest that disease mutations would lead to a range of defects in Gigaxonin stability, impairing its homodimerization, BTB or KELCH domain folding, or CUL3 and substrate binding. We further demonstrate that regardless of the mutations or the severity of the disease, Gigaxonin abundance is severely reduced in all GAN patients due to both mRNA and protein instability mechanisms.
Conclusions: In this study, we developed a new penetrant and specific test to diagnose GAN among a set of individuals exhibiting CMT2 of unknown etiology to suggest that the prevalence of GAN is probably under-evaluated among peripheral neuropathies. We propose to use this new test in concert with the clinical examination and prior to the systematic screening of GAN mutations that has shown strong limitations for large deletions. Combining the generation of the structural modeling of Gigaxonin to an analysis of Gigaxonin transcripts and proteins in patients, we provide the first evidences of the instability of this E3 ligase adaptor in disease.
Figures
References
-
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, Demir E, Topaloglu H, Korinthenberg R, Tuysuz B, Landrieu P, Hentati F, Koenig M. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26:370–374. doi: 10.1038/81701. - DOI - PubMed
-
- Berg BO, Rosenberg SH, Asbury AK. Giant axonal neuropathy. Pediatrics. 1972;49:894–899. - PubMed
-
- Azzedine H, Ravise N, Verny C, Gabreels-Festen A, Lammens M, Grid D, Vallat JM, Durosier G, Senderek J, Nouioua S, Hamadouche T, Bouhouche A, Guilbot A, Stendel C, Ruberg M, Brice A, Birouk N, Dubourg O, Tazir M, LeGuern E. Spine deformities in Charcot-Marie-Tooth 4C caused by SH3TC2 gene mutations. Neurology. 2006;67:602–606. doi: 10.1212/01.wnl.0000230225.19797.93. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
