

Acylcarnitines activate proinflammatory signaling pathways
- PMID: 24760988
- PMCID: PMC4059985
- DOI: 10.1152/ajpendo.00656.2013
Acylcarnitines activate proinflammatory signaling pathways
Abstract
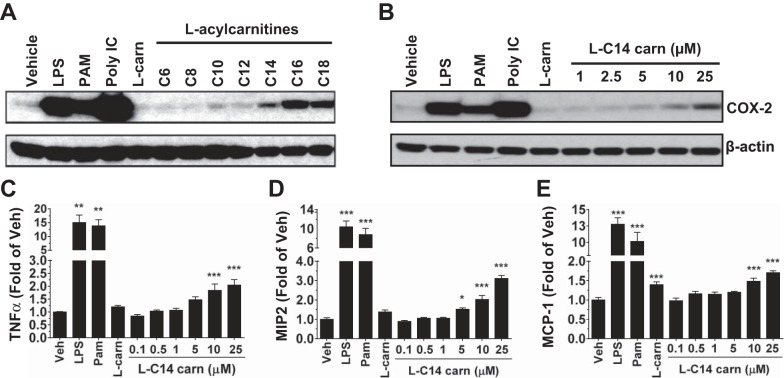
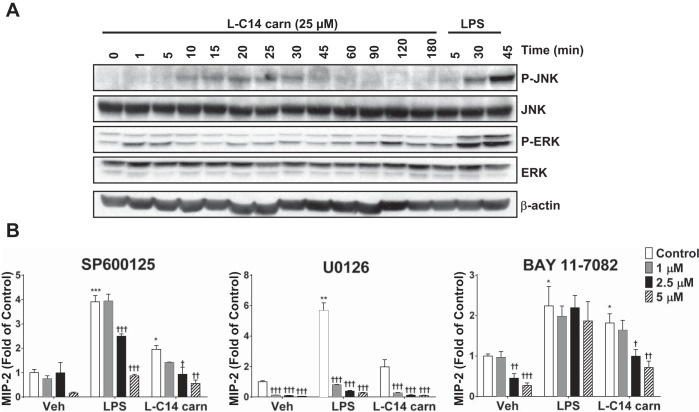
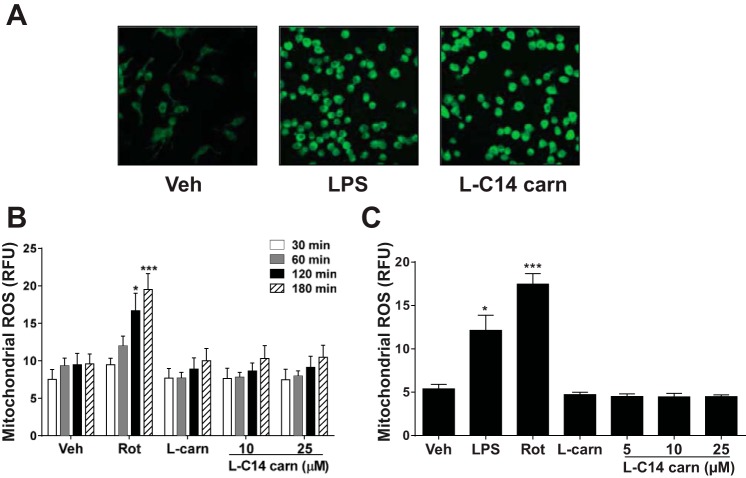
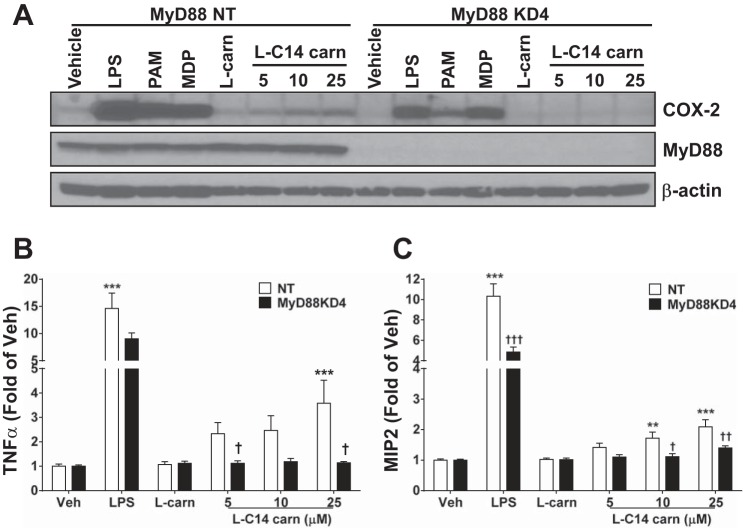
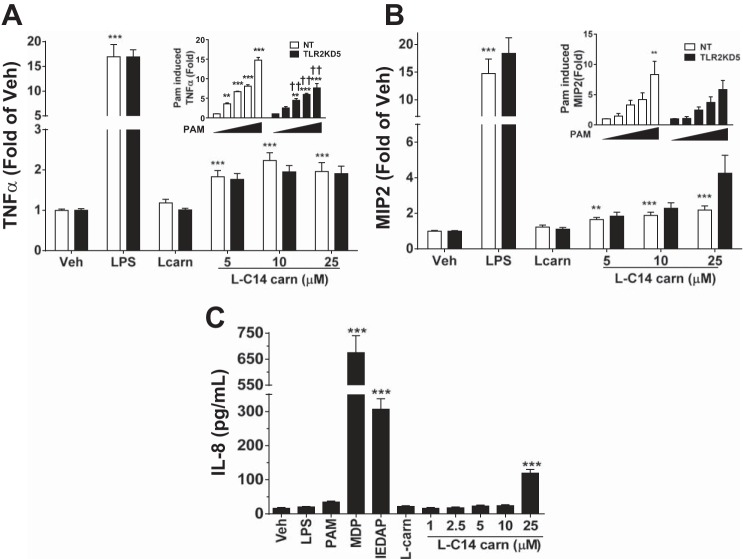
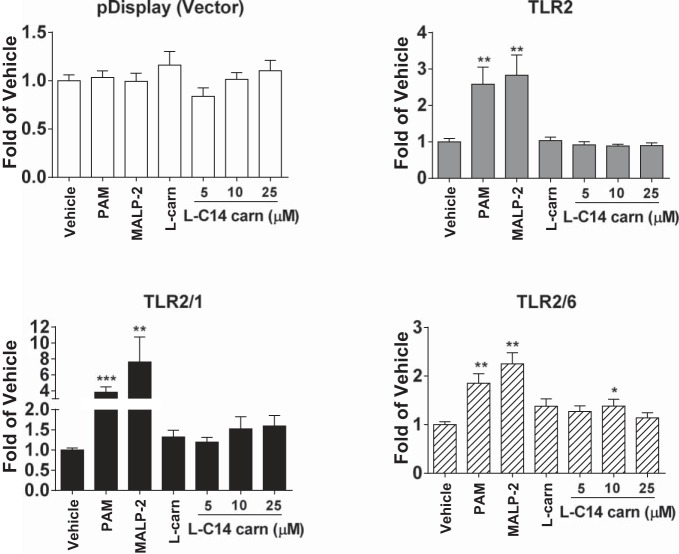
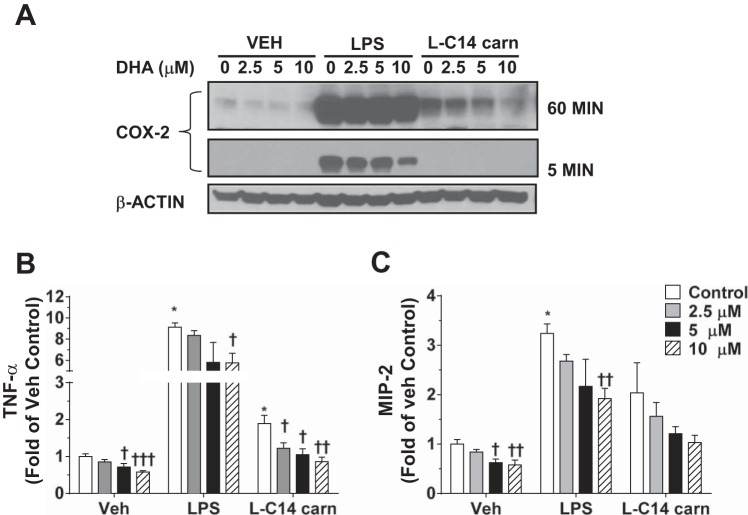
Incomplete β-oxidation of fatty acids in mitochondria is a feature of insulin resistance and type 2 diabetes mellitus (T2DM). Previous studies revealed that plasma concentrations of medium- and long-chain acylcarnitines (by-products of incomplete β-oxidation) are elevated in T2DM and insulin resistance. In a previous study, we reported that mixed D,L isomers of C12- or C14-carnitine induced an NF-κB-luciferase reporter gene in RAW 264.7 cells, suggesting potential activation of proinflammatory pathways. Here, we determined whether the physiologically relevant L-acylcarnitines activate classical proinflammatory signaling pathways and if these outcomes involve pattern recognition receptor (PRR)-associated pathways. Acylcarnitines induced the expression of cyclooxygenase-2 in a chain length-dependent manner in RAW 264.7 cells. L-C14 carnitine (5-25 μM), used as a representative acylcarnitine, stimulated the expression and secretion of proinflammatory cytokines in a dose-dependent manner. Furthermore, L-C14 carnitine induced phosphorylation of JNK and ERK, common downstream components of many proinflammatory signaling pathways including PRRs. Knockdown of MyD88, a key cofactor in PRR signaling and inflammation, blunted the proinflammatory effects of acylcarnitine. While these results point to potential involvement of PRRs, L-C14 carnitine promoted IL-8 secretion from human epithelial cells (HCT-116) lacking Toll-like receptors (TLR)2 and -4, and did not activate reporter constructs in TLR overexpression cell models. Thus, acylcarnitines have the potential to activate inflammation, but the specific molecular and tissue target(s) involved remain to be identified.
Keywords: TLR; acylcarnitine; inflammation; pattern recognition receptors; β-oxidation.
Figures
References
-
- Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW, Garvey WT. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr 139: 1073–1081, 2009 - PMC - PubMed
-
- Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw 17: 4–12, 2006 - PubMed
-
- De Vogel-van den Bosch J, Hoeks J, Timmers S, Houten SM, van Dijk PJ, Boon W, Van Beurden D, Schaart G, Kersten S, Voshol PJ, Wanders RJ, Hesselink MK, Schrauwen P. The effects of long- or medium-chain fat diets on glucose tolerance and myocellular content of lipid intermediates in rats. Obesity (Silver Spring) 19: 792–799, 2011 - PubMed
-
- Ho JK, Duclos RI, Jr, Hamilton JA. Interactions of acyl carnitines with model membranes: a (13)C-NMR study. J Lipid Res 43: 1429–1439, 2002 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous