

Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis
- PMID: 24762401
- PMCID: PMC4073442
- DOI: 10.1681/ASN.2014010117
Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis
Abstract
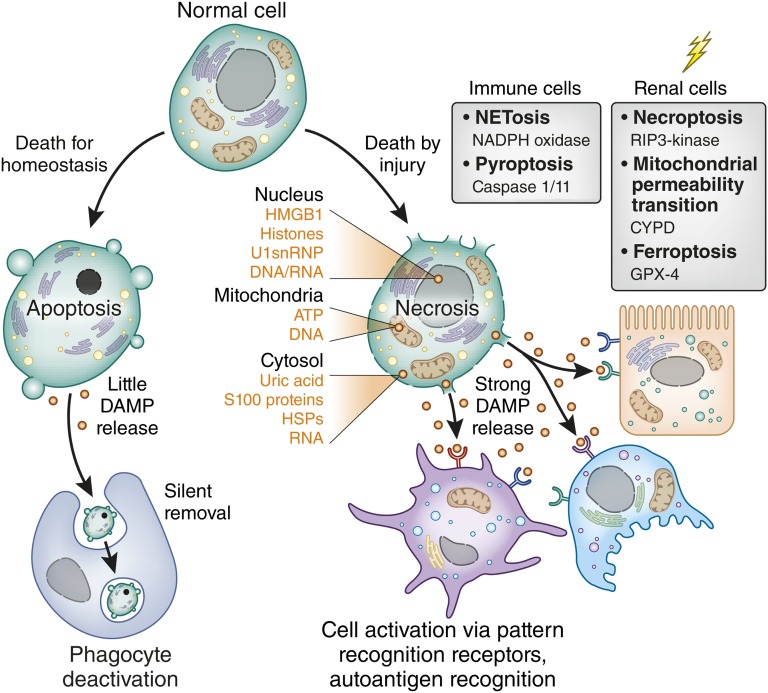
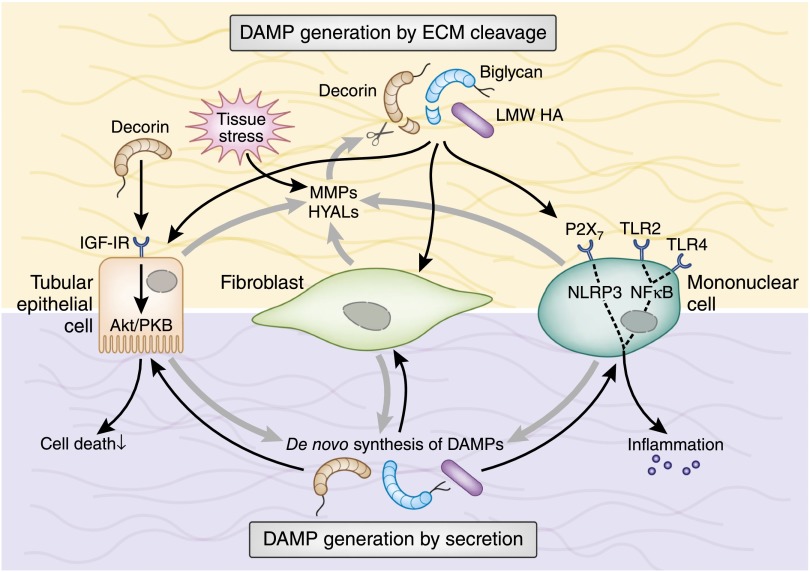
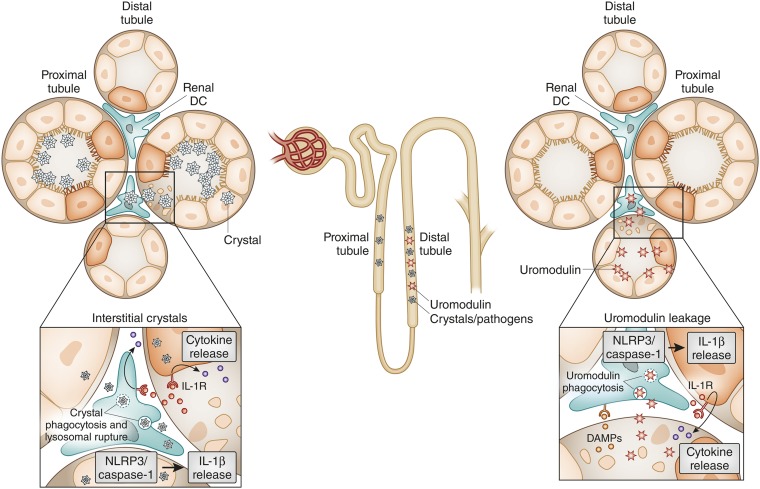
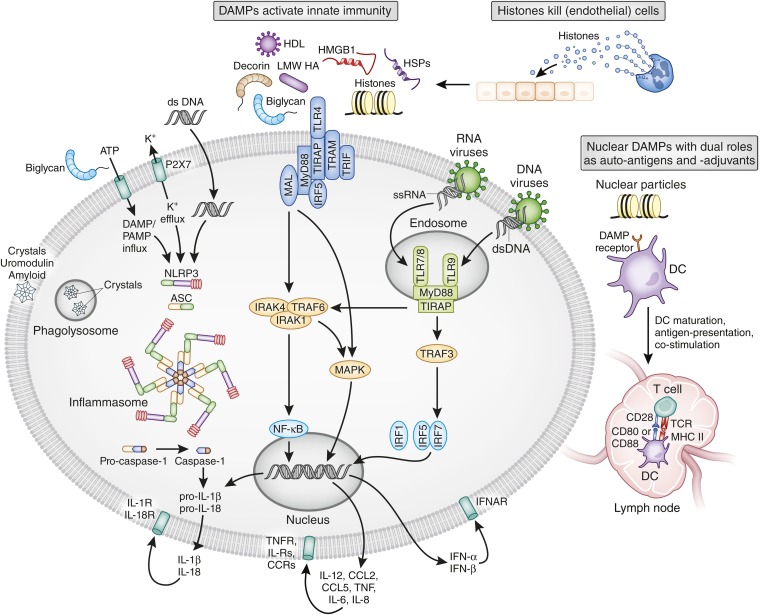
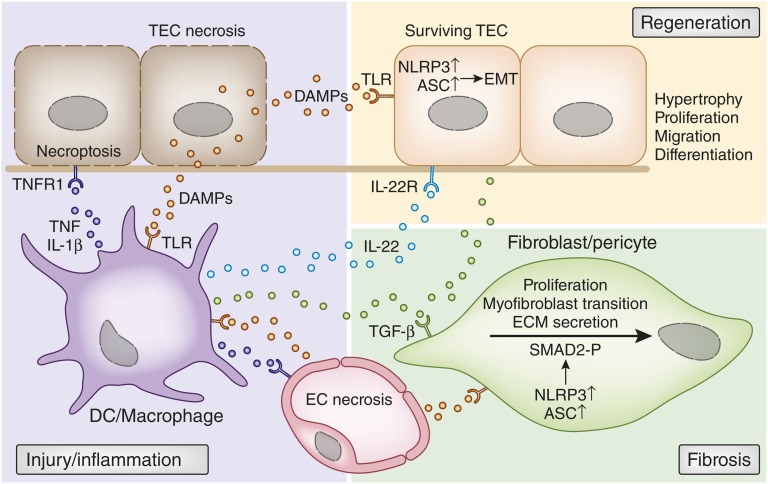
Tissue injury initiates an inflammatory response through the actions of immunostimulatory molecules referred to as damage-associated molecular patterns (DAMPs). DAMPs encompass a group of heterogenous molecules, including intracellular molecules released during cell necrosis and molecules involved in extracellular matrix remodeling such as hyaluronan, biglycan, and fibronectin. Kidney-specific DAMPs include crystals and uromodulin released by renal tubular damage. DAMPs trigger innate immunity by activating Toll-like receptors, purinergic receptors, or the NLRP3 inflammasome. However, recent evidence revealed that DAMPs also trigger re-epithelialization upon kidney injury and contribute to epithelial-mesenchymal transition and, potentially, to myofibroblast differentiation and proliferation. Thus, these discoveries suggest that DAMPs drive not only immune injury but also kidney regeneration and renal scarring. Here, we review the data from these studies and discuss the increasingly complex connection between DAMPs and kidney diseases.
Keywords: ARF; GN; immunology and pathology.
Copyright © 2014 by the American Society of Nephrology.
Figures
References
-
- Wallach D, Kang TB, Kovalenko A: Concepts of tissue injury and cell death in inflammation: A historical perspective. Nat Rev Immunol 14: 51–59, 2014 - PubMed
-
- Medzhitov R: Origin and physiological roles of inflammation. Nature 454: 428–435, 2008 - PubMed
-
- Matzinger P: Tolerance, danger, and the extended family. Annu Rev Immunol 12: 991–1045, 1994 - PubMed
-
- Kurts C, Panzer U, Anders HJ, Rees AJ: The immune system and kidney disease: Basic concepts and clinical implications. Nat Rev Immunol 13: 738–753, 2013 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
