

Quality control and conduct of genome-wide association meta-analyses
- PMID: 24762786
- PMCID: PMC4083217
- DOI: 10.1038/nprot.2014.071
Quality control and conduct of genome-wide association meta-analyses
Abstract
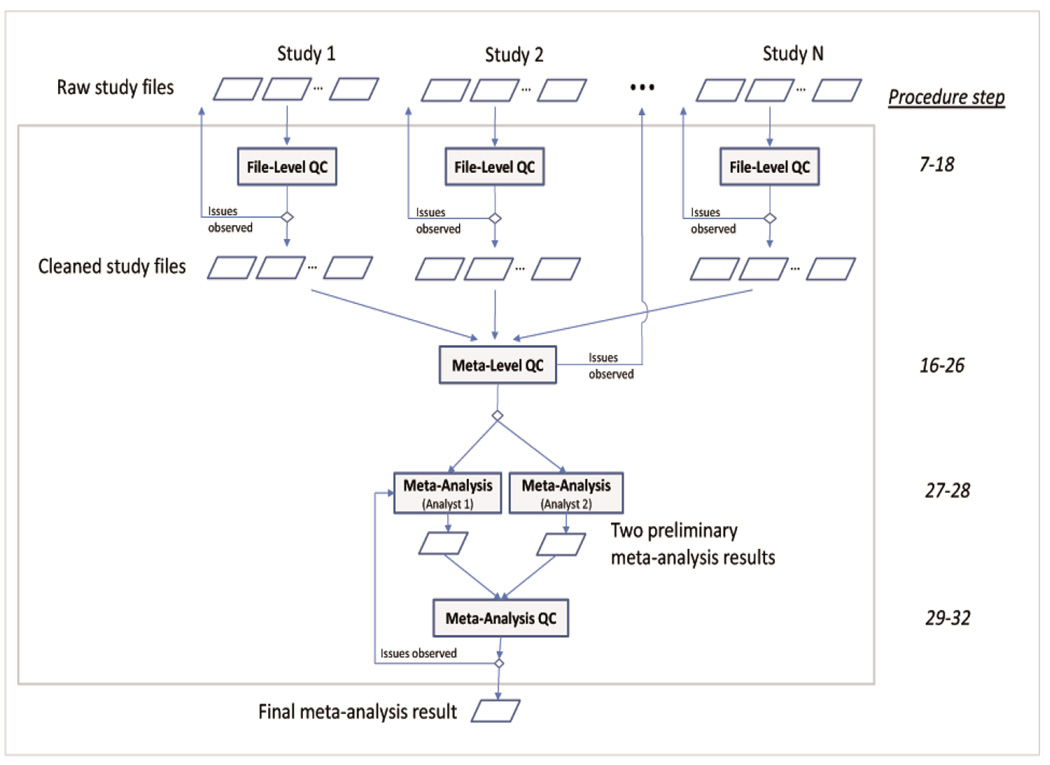
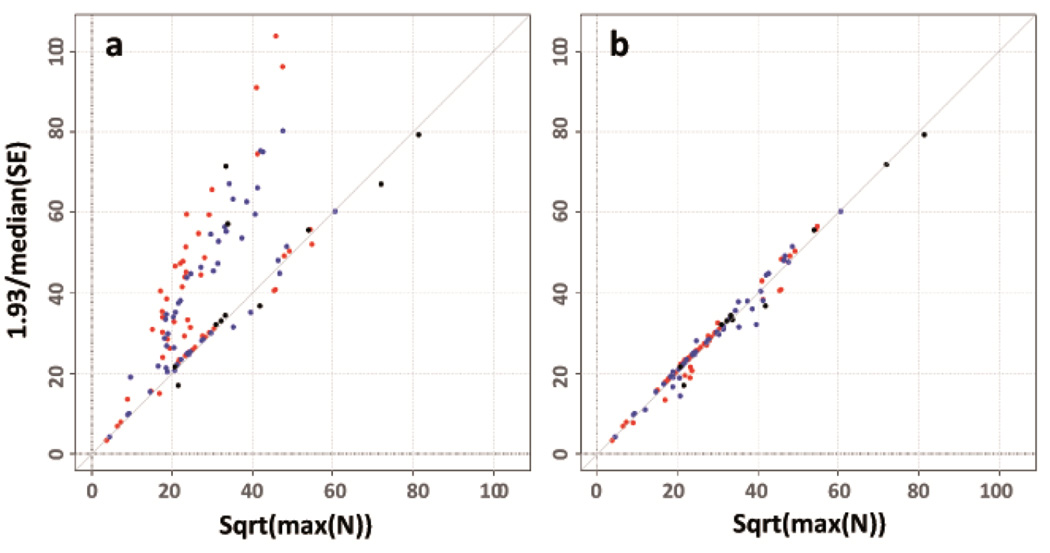
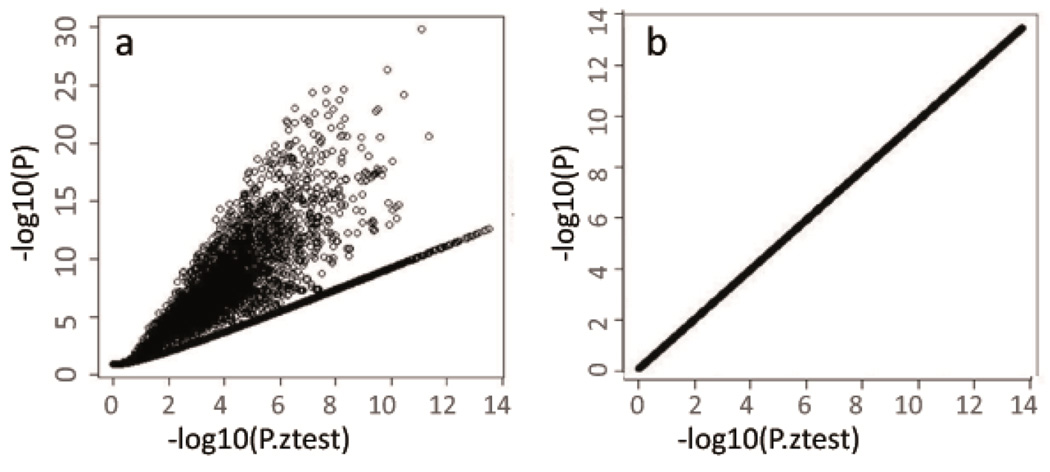
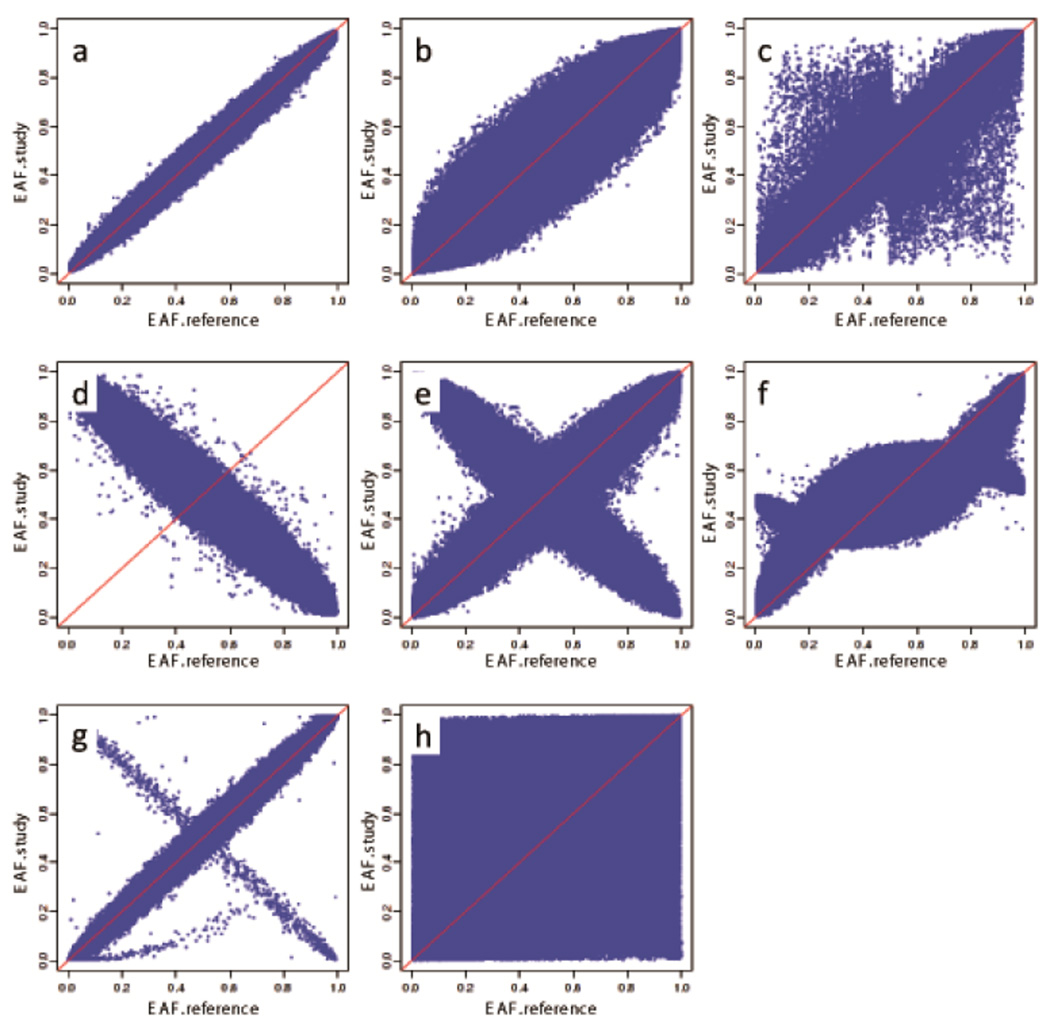
Rigorous organization and quality control (QC) are necessary to facilitate successful genome-wide association meta-analyses (GWAMAs) of statistics aggregated across multiple genome-wide association studies. This protocol provides guidelines for (i) organizational aspects of GWAMAs, and for (ii) QC at the study file level, the meta-level across studies and the meta-analysis output level. Real-world examples highlight issues experienced and solutions developed by the GIANT Consortium that has conducted meta-analyses including data from 125 studies comprising more than 330,000 individuals. We provide a general protocol for conducting GWAMAs and carrying out QC to minimize errors and to guarantee maximum use of the data. We also include details for the use of a powerful and flexible software package called EasyQC. Precise timings will be greatly influenced by consortium size. For consortia of comparable size to the GIANT Consortium, this protocol takes a minimum of about 10 months to complete.
Figures
References
-
- McCarthy MI, Hirschhorn JN. Genome-wide association studies: past, present and future. Human molecular genetics. 2008;17:R100–R101. - PubMed
-
- Hirschhorn JN, Gajdos ZK. Genome-wide association studies: results from the first few years and potential implications for clinical medicine. Annual review of medicine. 2011;62:11–24. - PubMed
Publication types
MeSH terms
Grants and funding
- DK078150/DK/NIDDK NIH HHS/United States
- CZB/4/672/CSO_/Chief Scientist Office/United Kingdom
- U106179472/MRC_/Medical Research Council/United Kingdom
- R01DK075787/DK/NIDDK NIH HHS/United States
- R01 DK075787/DK/NIDDK NIH HHS/United States
- MC_U106179471/MRC_/Medical Research Council/United Kingdom
- G0401527/MRC_/Medical Research Council/United Kingdom
- MR/K013351/1/MRC_/Medical Research Council/United Kingdom
- T32 HL007055/HL/NHLBI NIH HHS/United States
- R01 DK078150/DK/NIDDK NIH HHS/United States
- T32 HL007427/HL/NHLBI NIH HHS/United States
- G1000143/MRC_/Medical Research Council/United Kingdom
- MR/J012165/1/MRC_/Medical Research Council/United Kingdom
- U106179471/MRC_/Medical Research Council/United Kingdom
- MC_UU_12015/1/MRC_/Medical Research Council/United Kingdom
- R37 HL066289/HL/NHLBI NIH HHS/United States
- 097117/WT_/Wellcome Trust/United Kingdom
- MC_UU_12015/2/MRC_/Medical Research Council/United Kingdom
- 14136/CRUK_/Cancer Research UK/United Kingdom
- MC_U106179472/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
