

Integrated computational tools for identification of CCR5 antagonists as potential HIV-1 entry inhibitors: homology modeling, virtual screening, molecular dynamics simulations and 3D QSAR analysis
- PMID: 24762964
- PMCID: PMC6270745
- DOI: 10.3390/molecules19045243
Integrated computational tools for identification of CCR5 antagonists as potential HIV-1 entry inhibitors: homology modeling, virtual screening, molecular dynamics simulations and 3D QSAR analysis
Abstract
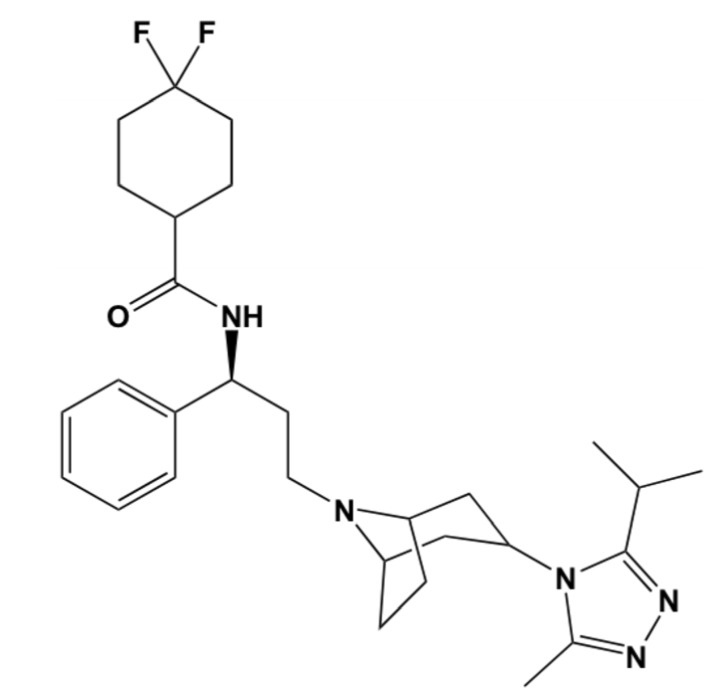
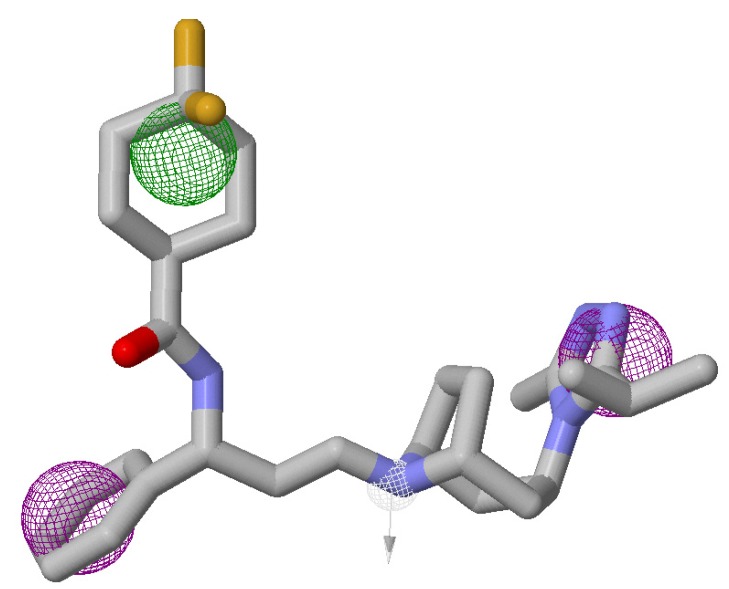
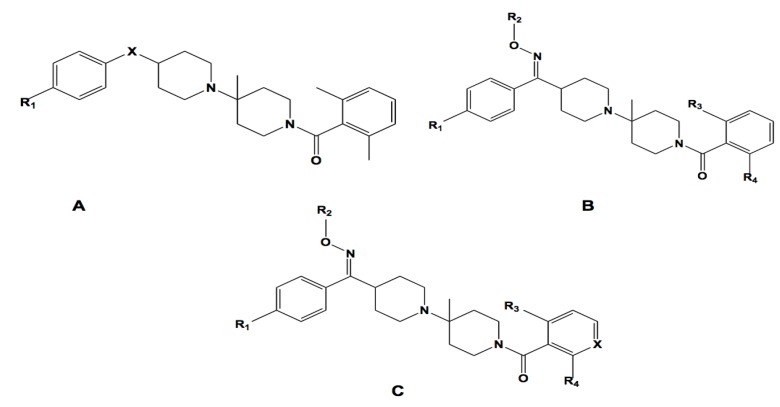
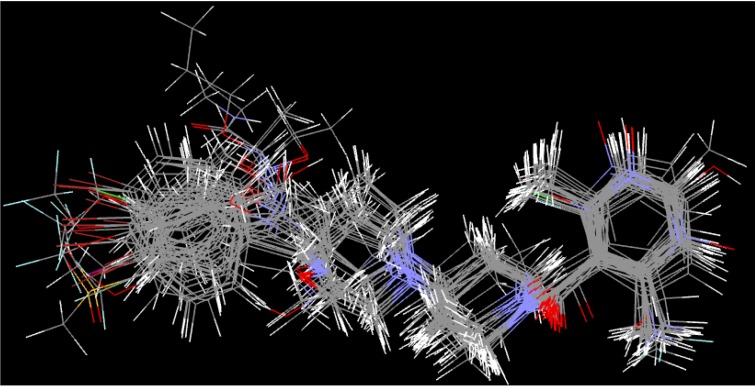
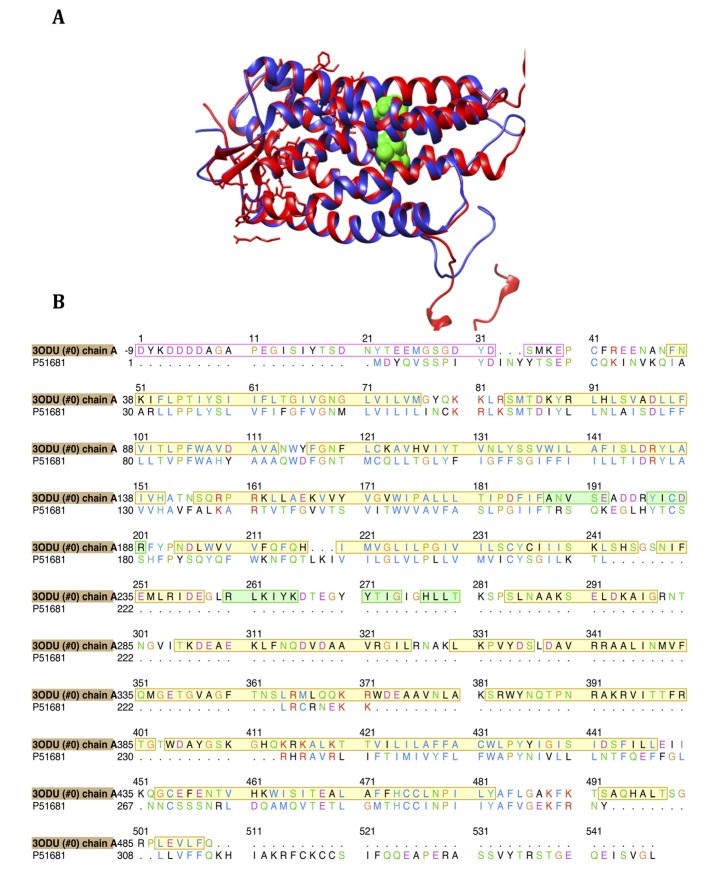
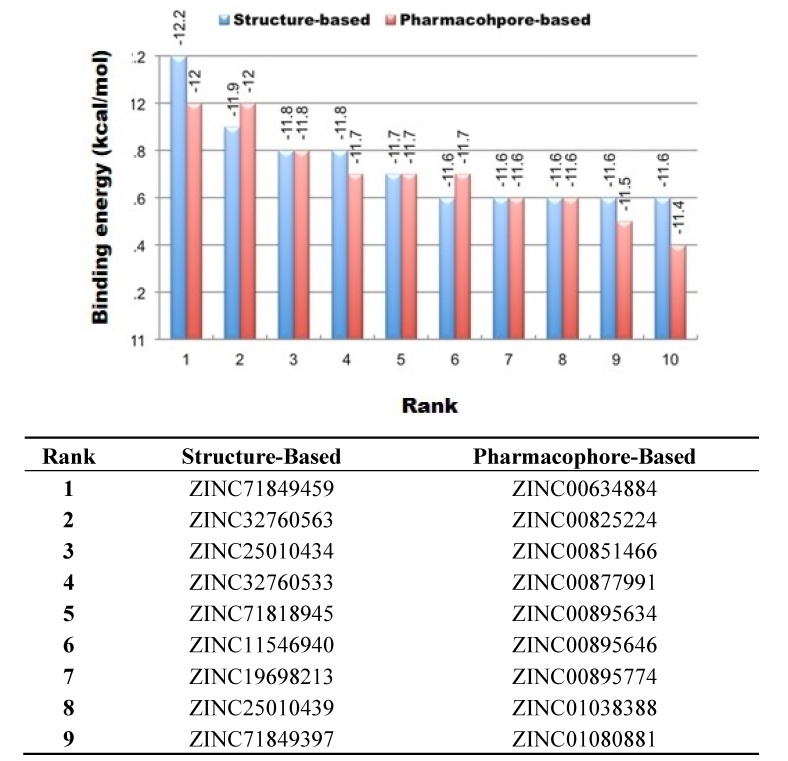
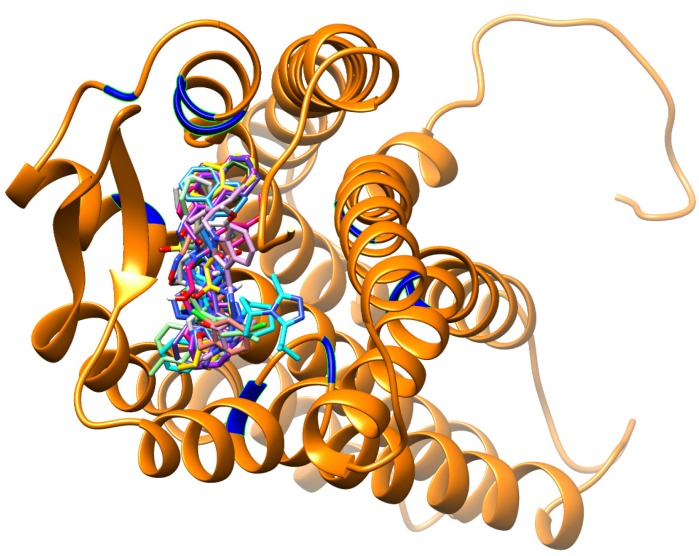
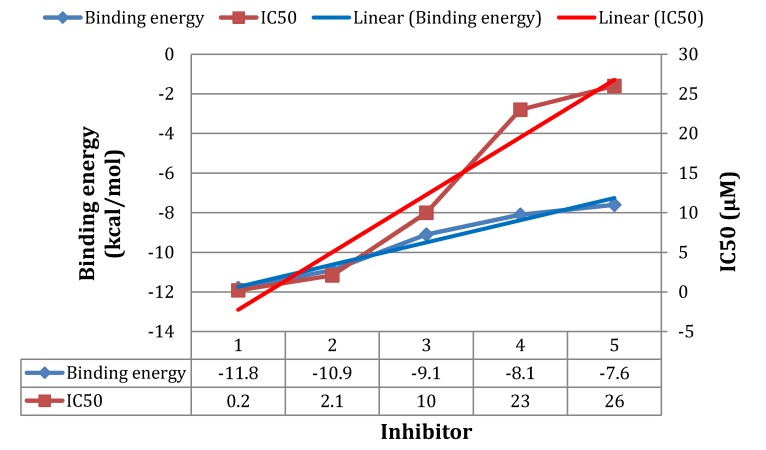
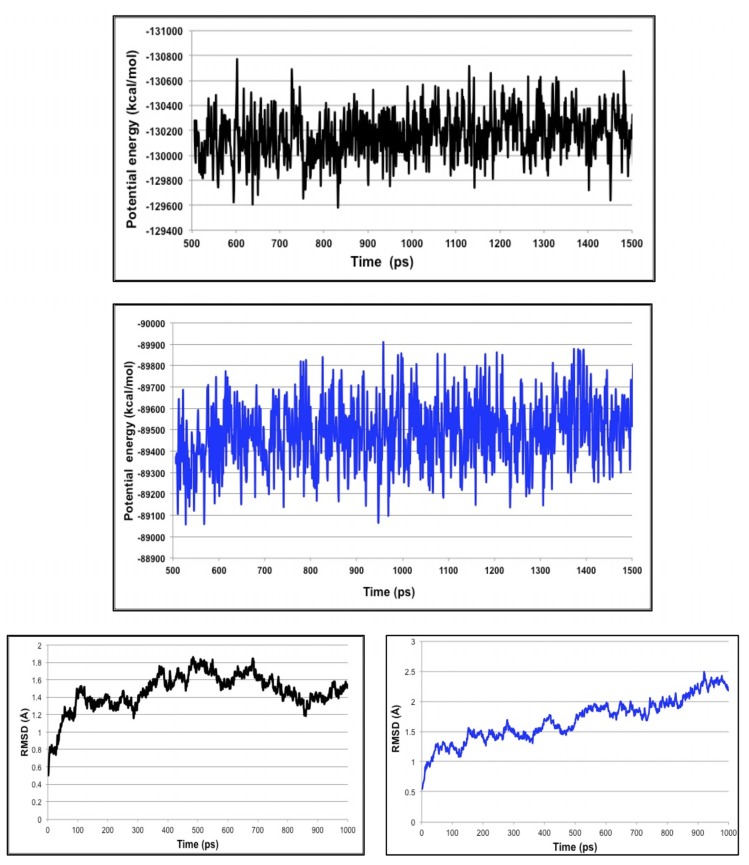
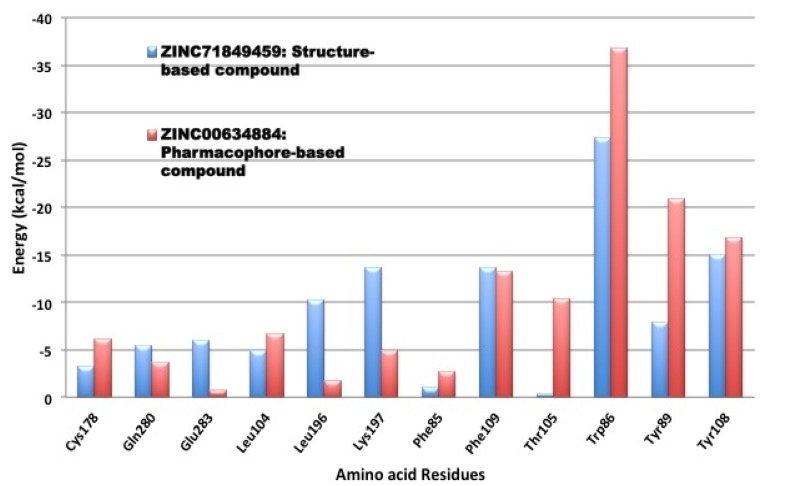
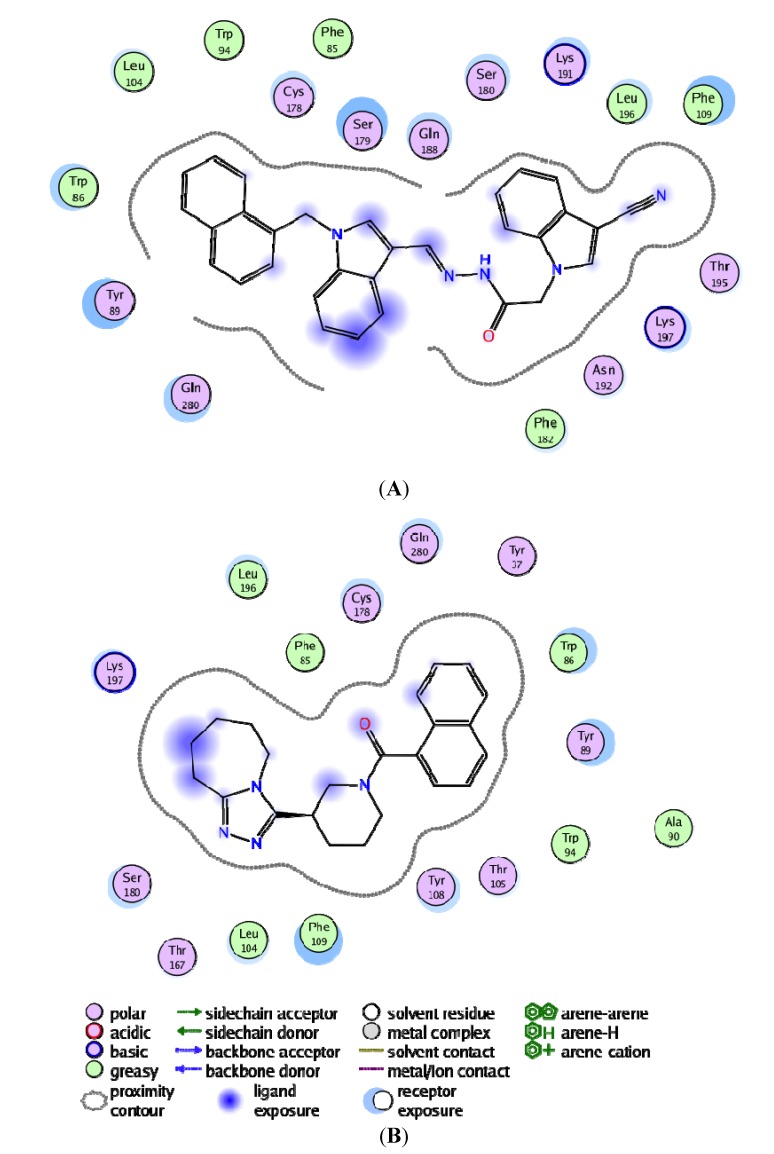
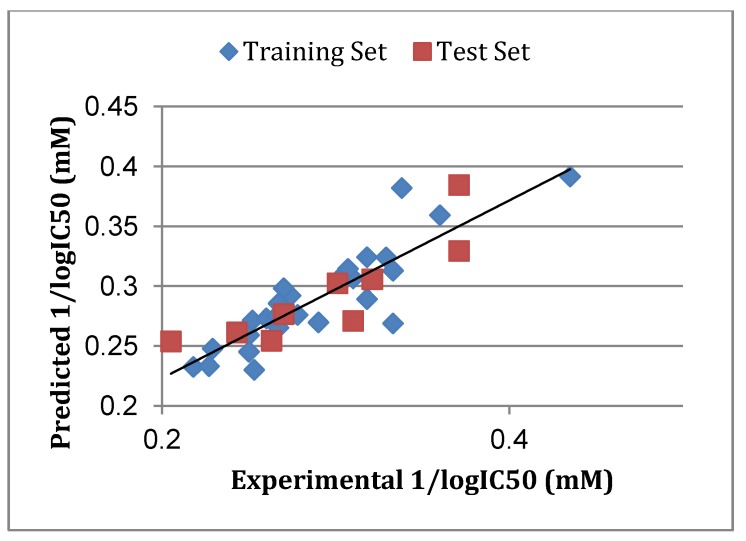
Using integrated in-silico computational techniques, including homology modeling, structure-based and pharmacophore-based virtual screening, molecular dynamic simulations, per-residue energy decomposition analysis and atom-based 3D-QSAR analysis, we proposed ten novel compounds as potential CCR5-dependent HIV-1 entry inhibitors. Via validated docking calculations, binding free energies revealed that novel leads demonstrated better binding affinities with CCR5 compared to maraviroc, an FDA-approved HIV-1 entry inhibitor and in clinical use. Per-residue interaction energy decomposition analysis on the averaged MD structure showed that hydrophobic active residues Trp86, Tyr89 and Tyr108 contributed the most to inhibitor binding. The validated 3D-QSAR model showed a high cross-validated rcv2 value of 0.84 using three principal components and non-cross-validated r2 value of 0.941. It was also revealed that almost all compounds in the test set and training set yielded a good predicted value. Information gained from this study could shed light on the activity of a new series of lead compounds as potential HIV entry inhibitors and serve as a powerful tool in the drug design and development machinery.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gadhe C.G., Kothandan G., Cho S.J. Computational modeling of human coreceptor CCR5 antagonist as a HIV-1 entry inhibitor: Using an integrated homology modeling, docking, and membrane molecular dynamics simulation analysis approach. J. Biomol. Struct. Dyn. 2013;31:1251–1279. doi: 10.1080/07391102.2012.732342. - DOI - PubMed
-
- Xu Y., Liu H., Niu C.Y., Luo C., Luo X.M., Shen J.H., Chen K.X., Jiang H.L. Molecular docking and 3D QSAR studies on 1-amino-2-phenyl-4-(piperidin-1-yl)-butanes based on the structural modeling of human CCR5 receptor. Bioorgan. Med. Chem. 2004;12:6193–6208. doi: 10.1016/j.bmc.2004.08.045. - DOI - PubMed
-
- Soliman M.E.S. A Hybrid Structure/Pharmacophore-Based Virtual Screening Approach to Design Potential Leads: A Computer-Aided Design of South African HIV-1 Subtype C Protease Inhibitors. Drug Dev. Res. 2013;74:283–295. doi: 10.1002/ddr.21078. - DOI
-
- Johnson B.C., Pauly G.T., Rai G., Patel D., Bauman J.D., Baker H.L., Das K., Schneider J.P., Maloney D.J., Arnold E., et al. A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology. 2012;9:1–23. doi: 10.1186/1742-4690-9-1. - DOI - PMC - PubMed
-
- Patel J.R., Prajapati L.M. Predictive QSAR modeling on tetrahydropyrimidine-2-one derivatives as HIV-1 protease enzyme inhibitors. Med. Chem. Res. 2013;22:2795–2801. doi: 10.1007/s00044-012-0275-8. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
