

Discrete molecular dynamics can predict helical prestructured motifs in disordered proteins
- PMID: 24763499
- PMCID: PMC3998973
- DOI: 10.1371/journal.pone.0095795
Discrete molecular dynamics can predict helical prestructured motifs in disordered proteins
Abstract
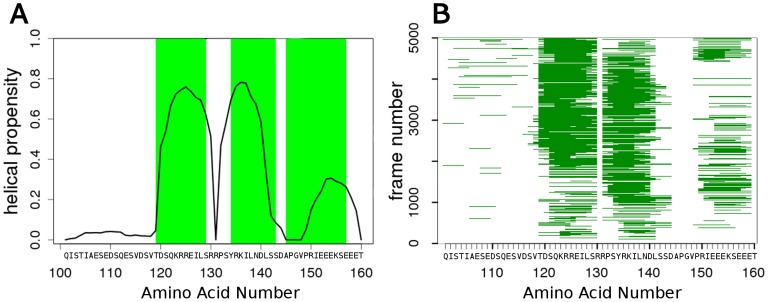
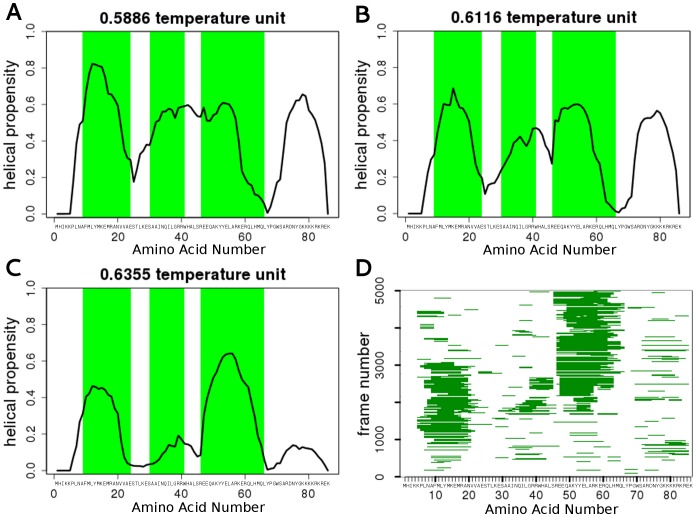
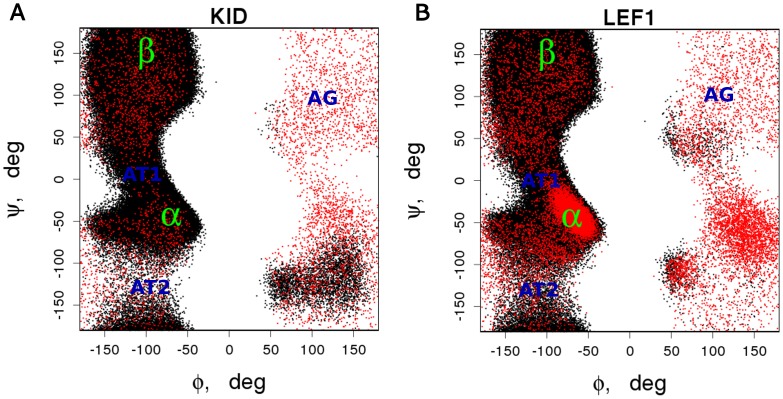
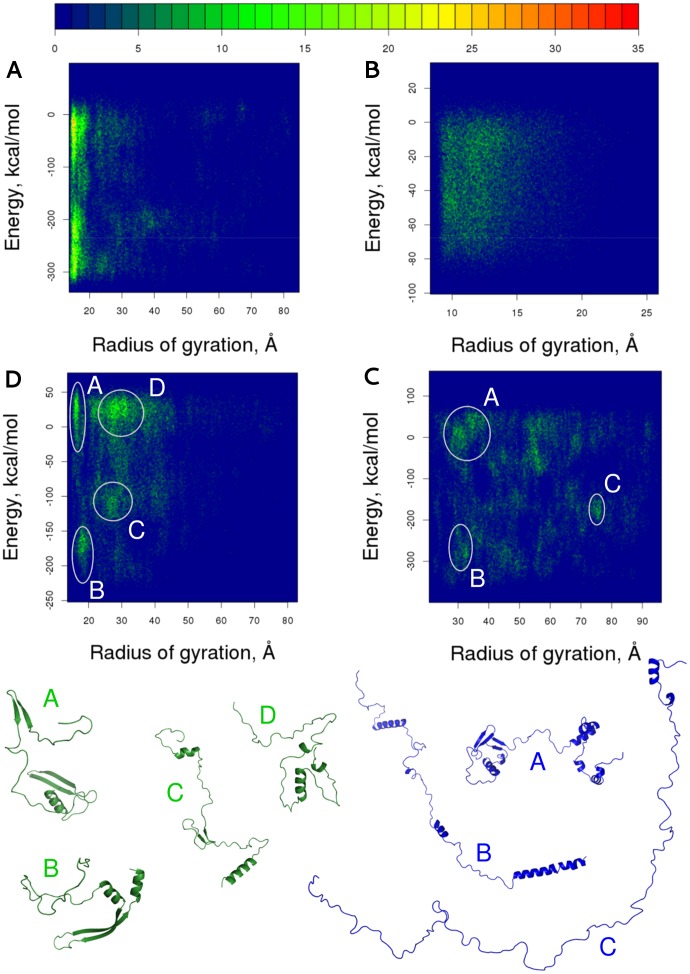
Intrinsically disordered proteins (IDPs) lack a stable tertiary structure, but their short binding regions termed Pre-Structured Motifs (PreSMo) can form transient secondary structure elements in solution. Although disordered proteins are crucial in many biological processes and designing strategies to modulate their function is highly important, both experimental and computational tools to describe their conformational ensembles and the initial steps of folding are sparse. Here we report that discrete molecular dynamics (DMD) simulations combined with replica exchange (RX) method efficiently samples the conformational space and detects regions populating α-helical conformational states in disordered protein regions. While the available computational methods predict secondary structural propensities in IDPs based on the observation of protein-protein interactions, our ab initio method rests on physical principles of protein folding and dynamics. We show that RX-DMD predicts α-PreSMos with high confidence confirmed by comparison to experimental NMR data. Moreover, the method also can dissect α-PreSMos in close vicinity to each other and indicate helix stability. Importantly, simulations with disordered regions forming helices in X-ray structures of complexes indicate that a preformed helix is frequently the binding element itself, while in other cases it may have a role in initiating the binding process. Our results indicate that RX-DMD provides a breakthrough in the structural and dynamical characterization of disordered proteins by generating the structural ensembles of IDPs even when experimental data are not available.
Conflict of interest statement
Figures
References
-
- Dunker AK, Silman I, Uversky VN, Sussman JL (2008) Function and structure of inherently disordered proteins. Curr Opin Struct Biol 18: 756–764. - PubMed
-
- Tompa P (2011) Unstructural biology coming of age. Curr Opin Struct Biol 21: 419–425. - PubMed
-
- Meszaros B, Simon I, Dosztanyi Z (2011) The expanding view of protein-protein interactions: complexes involving intrinsically disordered proteins. Phys Biol 8: 035003. - PubMed
-
- Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN (2009) Unfoldomics of human genetic diseases: illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept Lett 16: 1533–1547. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
