

Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function
- PMID: 24766809
- PMCID: PMC4146440
- DOI: 10.1016/j.cell.2014.02.058
Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function
Abstract
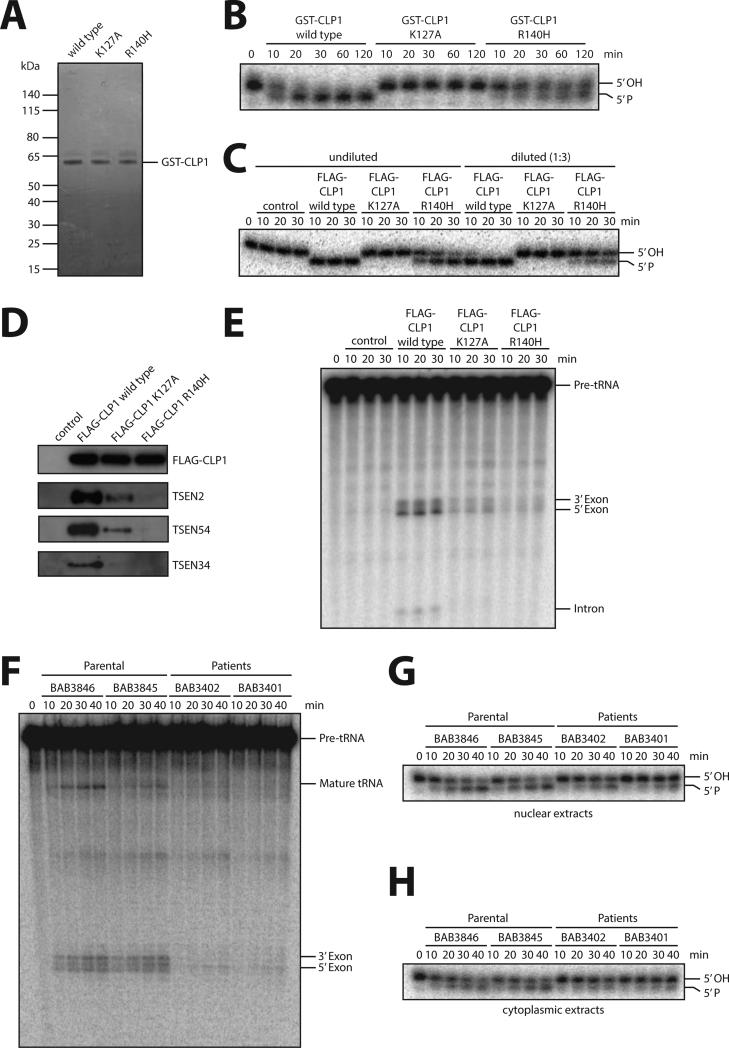
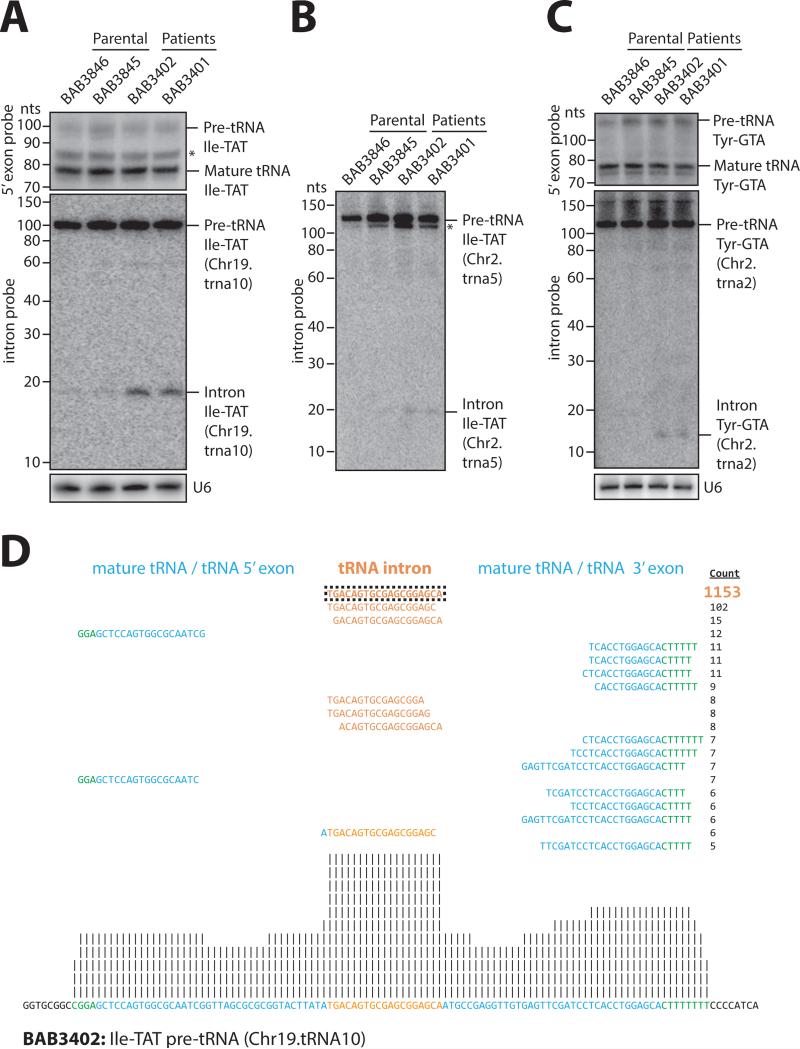
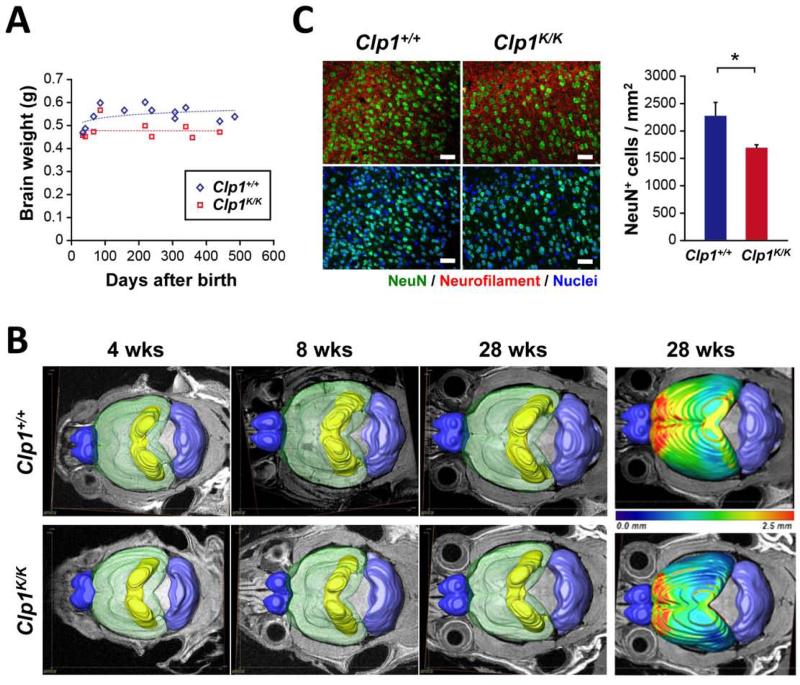
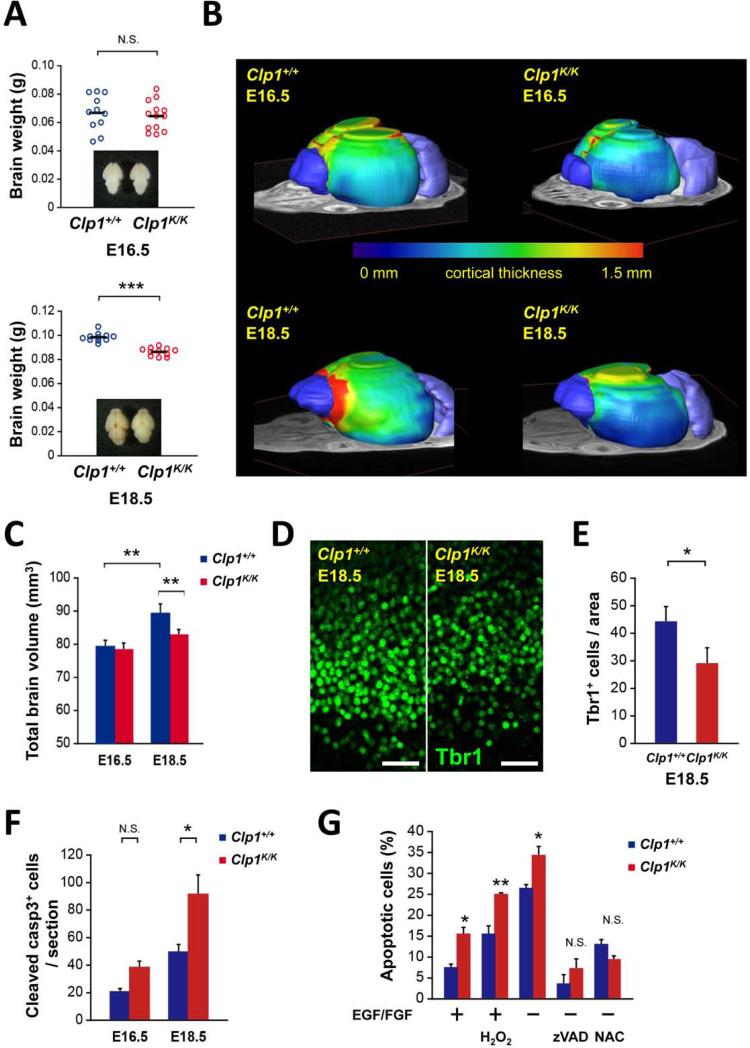
CLP1 is a RNA kinase involved in tRNA splicing. Recently, CLP1 kinase-dead mice were shown to display a neuromuscular disorder with loss of motor neurons and muscle paralysis. Human genome analyses now identified a CLP1 homozygous missense mutation (p.R140H) in five unrelated families, leading to a loss of CLP1 interaction with the tRNA splicing endonuclease (TSEN) complex, largely reduced pre-tRNA cleavage activity, and accumulation of linear tRNA introns. The affected individuals develop severe motor-sensory defects, cortical dysgenesis, and microcephaly. Mice carrying kinase-dead CLP1 also displayed microcephaly and reduced cortical brain volume due to the enhanced cell death of neuronal progenitors that is associated with reduced numbers of cortical neurons. Our data elucidate a neurological syndrome defined by CLP1 mutations that impair tRNA splicing. Reduction of a founder mutation to homozygosity illustrates the importance of rare variations in disease and supports the clan genomics hypothesis.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures



References
-
- Bayat V, Thiffault I, Jaiswal M, Tetreault M, Donti T, Sasarman F, Bernard G, Demers-Lamarche J, Dicaire MJ, Mathieu J, et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. - PMC - PubMed
-
- Budde BS, Namavar Y, Barth PG, Poll-The BT, Nurnberg G, Becker C, van Ruissen F, Weterman MA, Fluiter K, te Beek ET, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40:1113–1118. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
