

Simulation of Finnish population history, guided by empirical genetic data, to assess power of rare-variant tests in Finland
- PMID: 24768551
- PMCID: PMC4067550
- DOI: 10.1016/j.ajhg.2014.03.019
Simulation of Finnish population history, guided by empirical genetic data, to assess power of rare-variant tests in Finland
Abstract
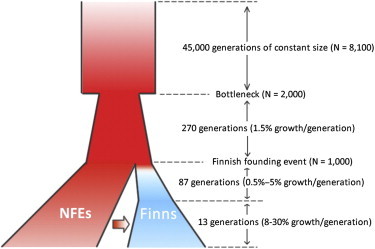
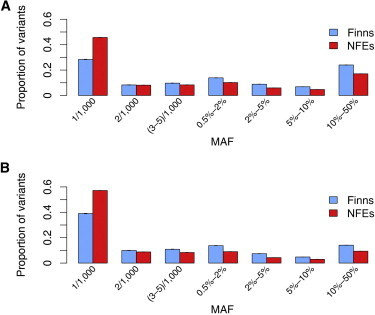
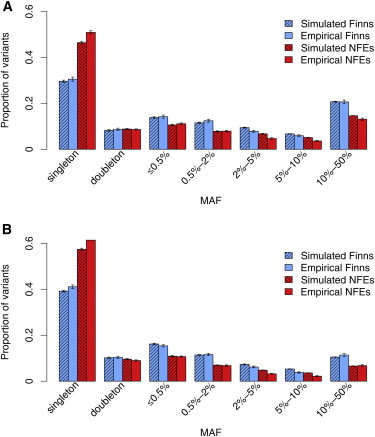
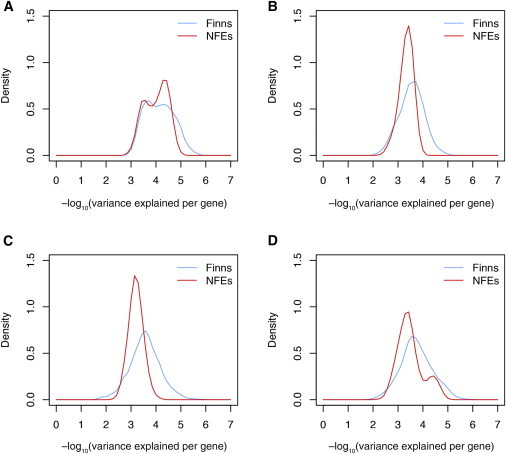
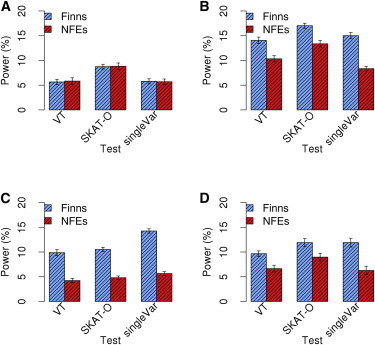
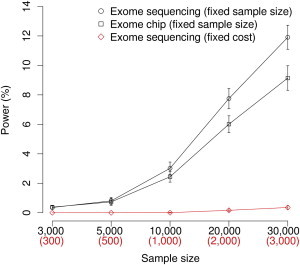
Finnish samples have been extensively utilized in studying single-gene disorders, where the founder effect has clearly aided in discovery, and more recently in genome-wide association studies of complex traits, where the founder effect has had less obvious impacts. As the field starts to explore rare variants' contribution to polygenic traits, it is of great importance to characterize and confirm the Finnish founder effect in sequencing data and to assess its implications for rare-variant association studies. Here, we employ forward simulation, guided by empirical deep resequencing data, to model the genetic architecture of quantitative polygenic traits in both the general European and the Finnish populations simultaneously. We demonstrate that power of rare-variant association tests is higher in the Finnish population, especially when variants' phenotypic effects are tightly coupled with fitness effects and therefore reflect a greater contribution of rarer variants. SKAT-O, variable-threshold tests, and single-variant tests are more powerful than other rare-variant methods in the Finnish population across a range of genetic models. We also compare the relative power and efficiency of exome array genotyping to those of high-coverage exome sequencing. At a fixed cost, less expensive genotyping strategies have far greater power than sequencing; in a fixed number of samples, however, genotyping arrays miss a substantial portion of genetic signals detected in sequencing, even in the Finnish founder population. As genetic studies probe sequence variation at greater depth in more diverse populations, our simulation approach provides a framework for evaluating various study designs for gene discovery.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
- R01 DK098032/DK/NIDDK NIH HHS/United States
- F32 GM106656/GM/NIGMS NIH HHS/United States
- T32 HG000040/HG/NHGRI NIH HHS/United States
- F32GM106656/GM/NIGMS NIH HHS/United States
- T32GM007748-33/GM/NIGMS NIH HHS/United States
- T32 GM008313/GM/NIGMS NIH HHS/United States
- 2R01DK075787/DK/NIDDK NIH HHS/United States
- T32GM007753/GM/NIGMS NIH HHS/United States
- T32 GM007748/GM/NIGMS NIH HHS/United States
- 1RC2DK088389-01/DK/NIDDK NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R01 DK075787/DK/NIDDK NIH HHS/United States
- RC2 DK088389/DK/NIDDK NIH HHS/United States
- T32GM008313/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
