

ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson's disease
- PMID: 24768991
- PMCID: PMC4134365
- DOI: 10.1016/j.mito.2014.04.008
ERK-mediated phosphorylation of TFAM downregulates mitochondrial transcription: implications for Parkinson's disease
Abstract
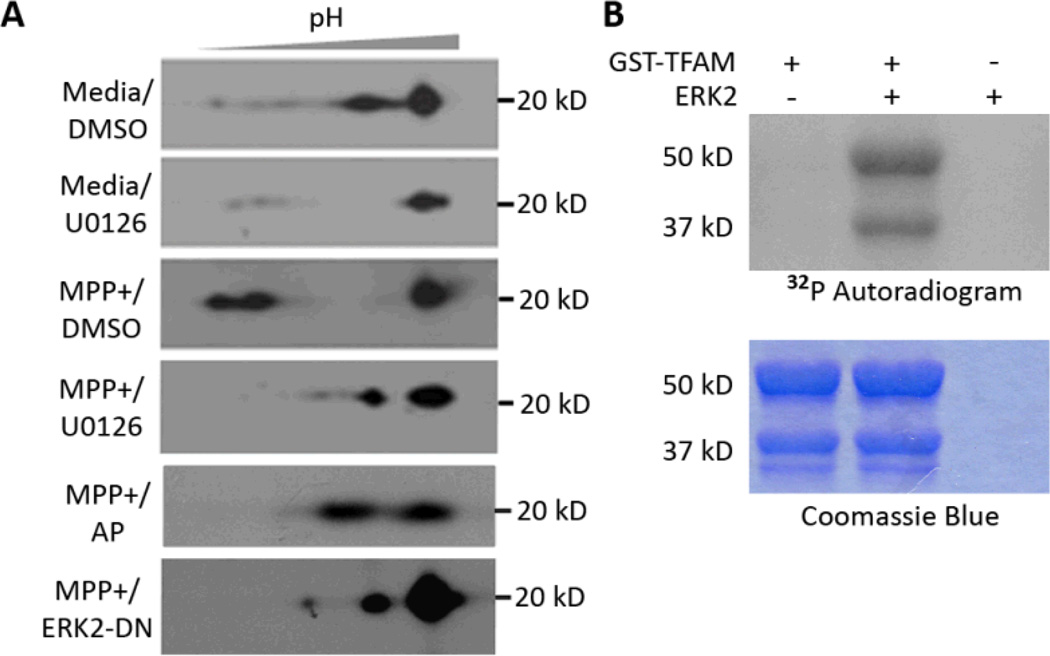
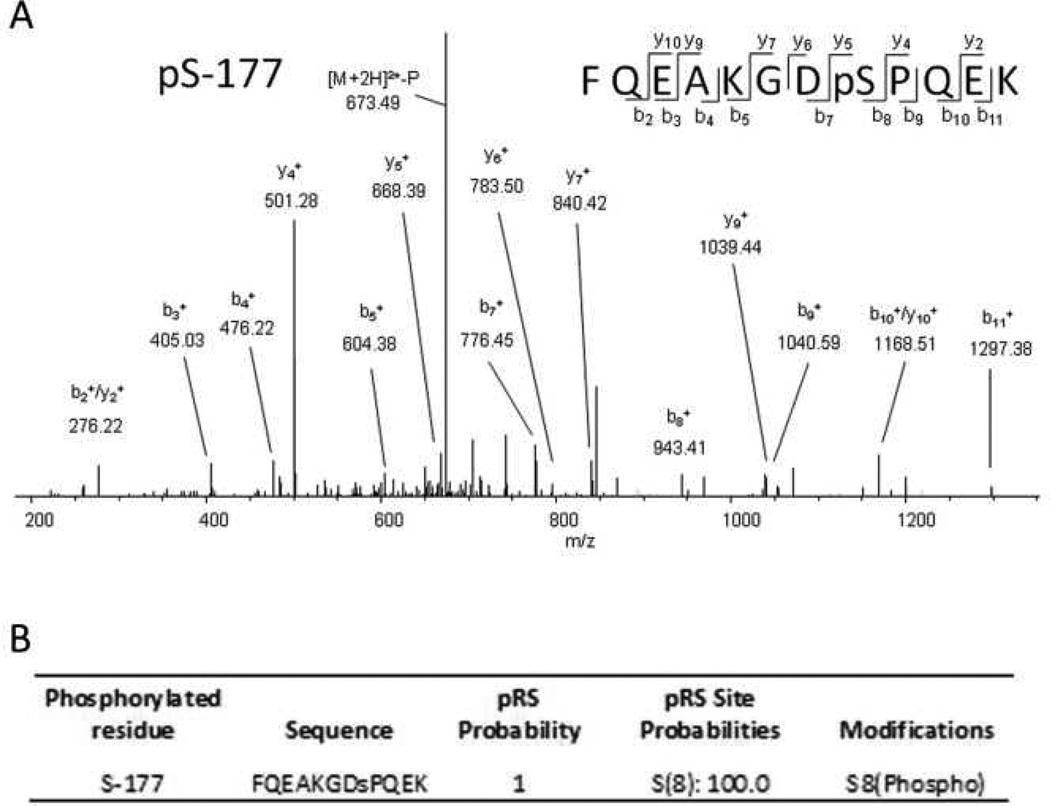
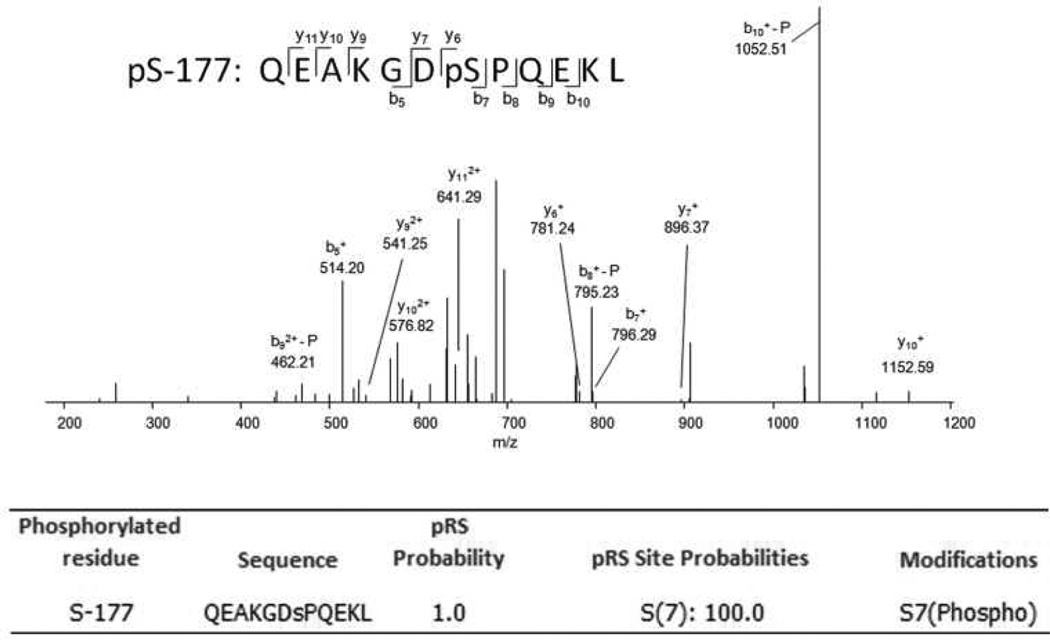
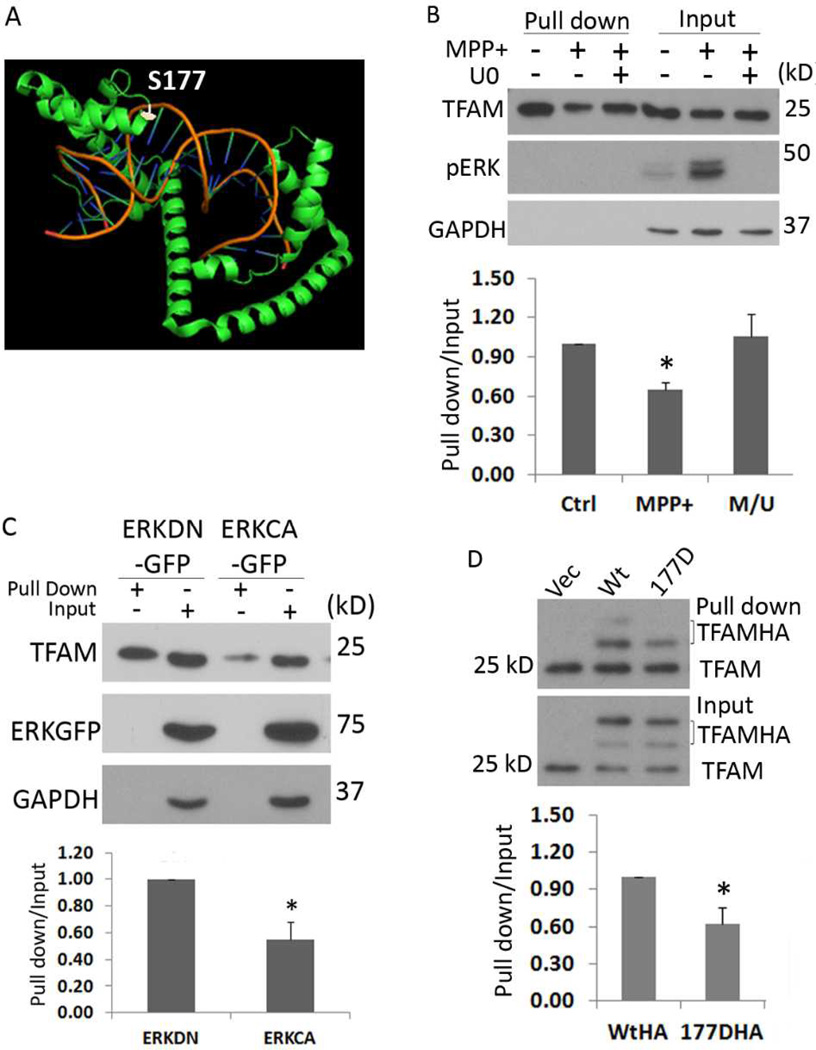
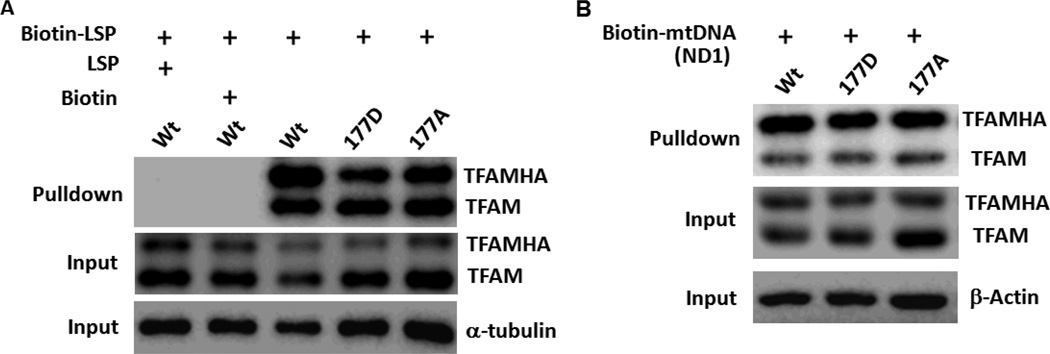
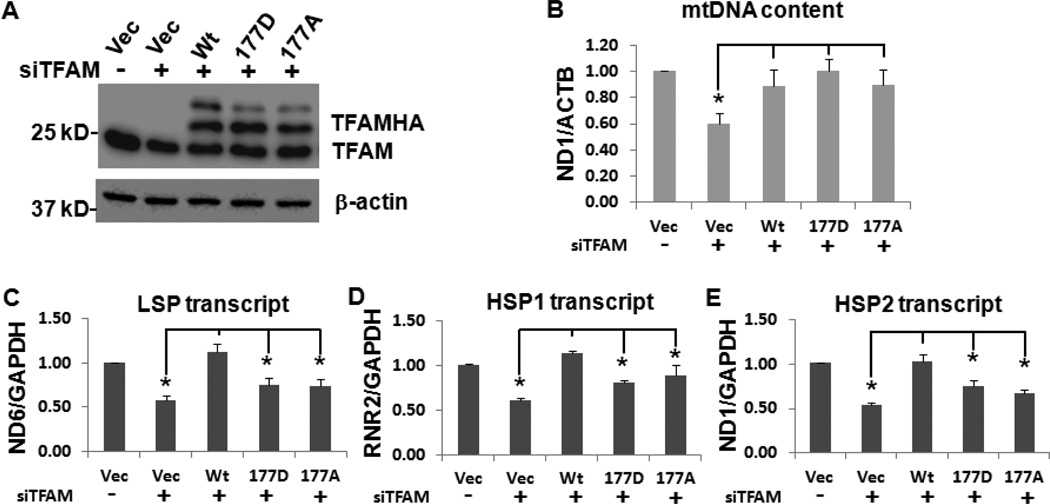
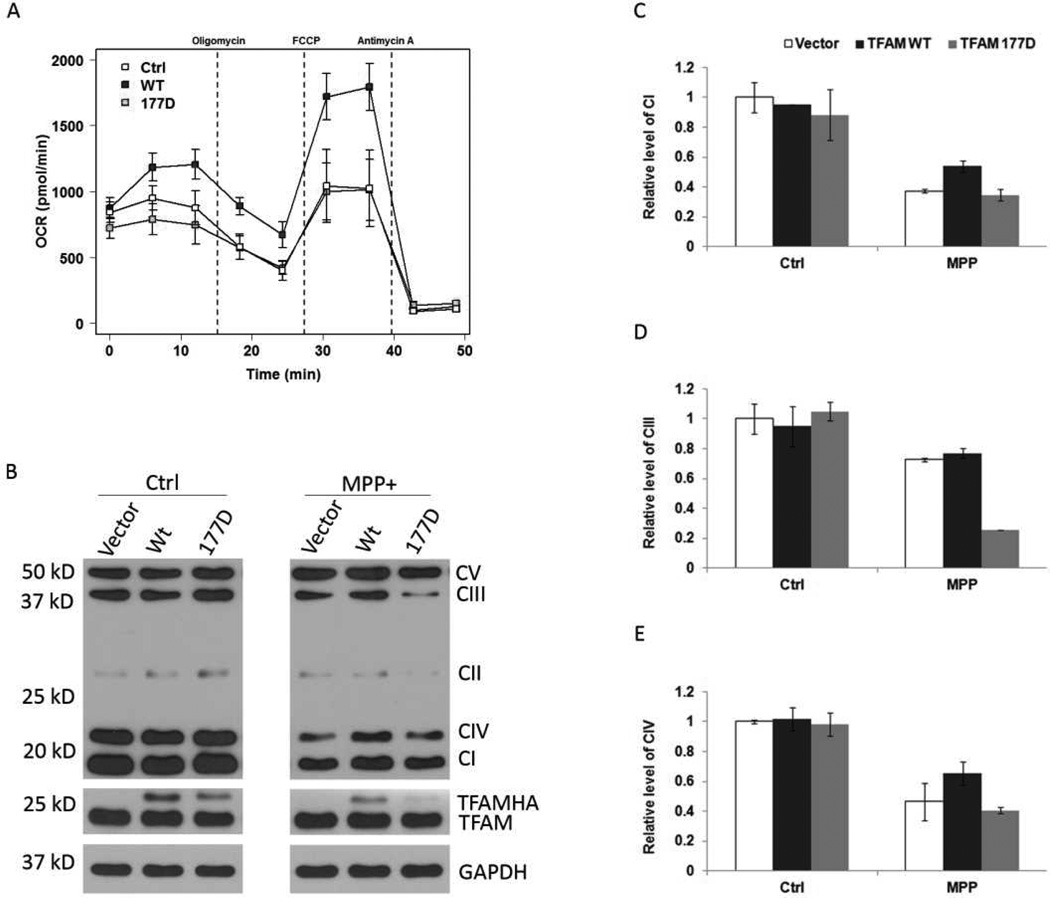
Mitochondrial transcription factor A (TFAM) regulates mitochondrial biogenesis, which is downregulated by extracellular signal-regulated protein kinases (ERK1/2) in cells treated chronically with the complex I inhibitor 1-methyl-4-phenylpyridinium (MPP+). We utilized mass spectrometry to identify ERK1/2-dependent TFAM phosphorylation sites. Mutation of TFAM at serine 177 to mimic phosphorylation recapitulated the effects of MPP+ in decreasing the binding of TFAM to the light strand promoter, suppressing mitochondrial transcription. Mutant TFAM was unable to affect respiratory function or rescue the effects of MPP+ on respiratory complexes. These data disclose a novel mechanism by which ERK1/2 regulates mitochondrial function through direct phosphorylation of TFAM.
Keywords: MAP kinases; Mitochondrial biogenesis; Parkinson disease; Phosphorylation; mtDNA.
Copyright © 2014 Elsevier B.V. and Mitochondria Research Society. All rights reserved.
Figures

References
-
- Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24:813–825. - PubMed
-
- Dairaghi DJ, Shadel GS, Clayton DA. Addition of a 29 residue carboxyl-terminal tail converts a simple HMG box-containing protein into a transcriptional activator. J Mol Biol. 1995;249:11–28. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
