

GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development
- PMID: 24770533
- PMCID: PMC4015328
- DOI: 10.1038/ncomms4680
GATA-dependent regulatory switches establish atrioventricular canal specificity during heart development
Abstract
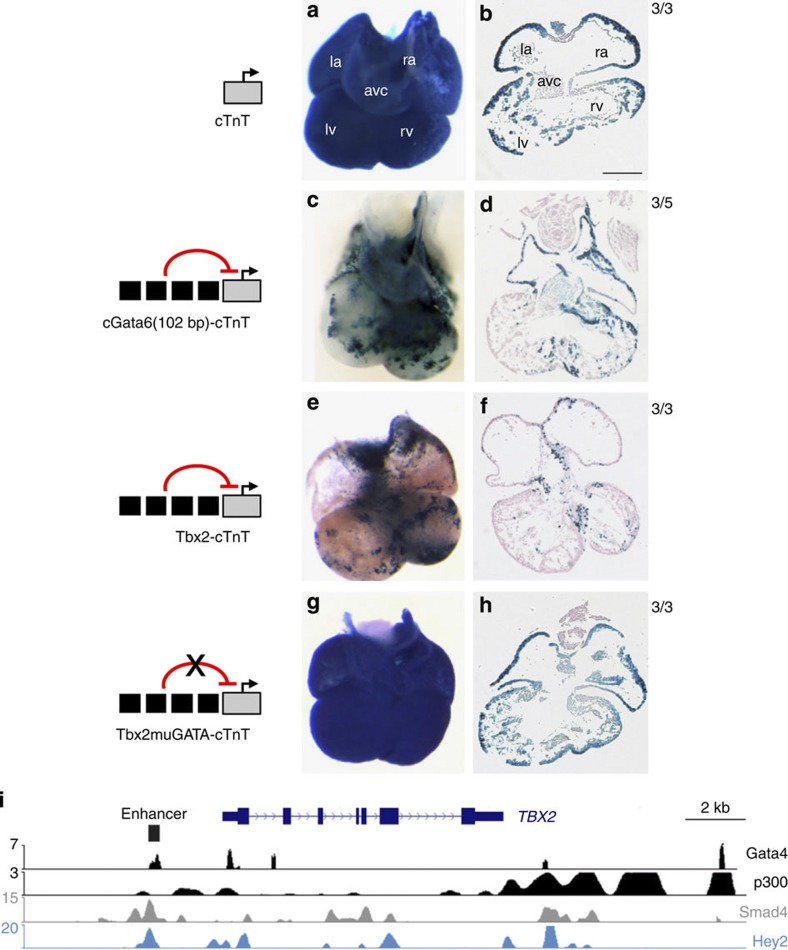
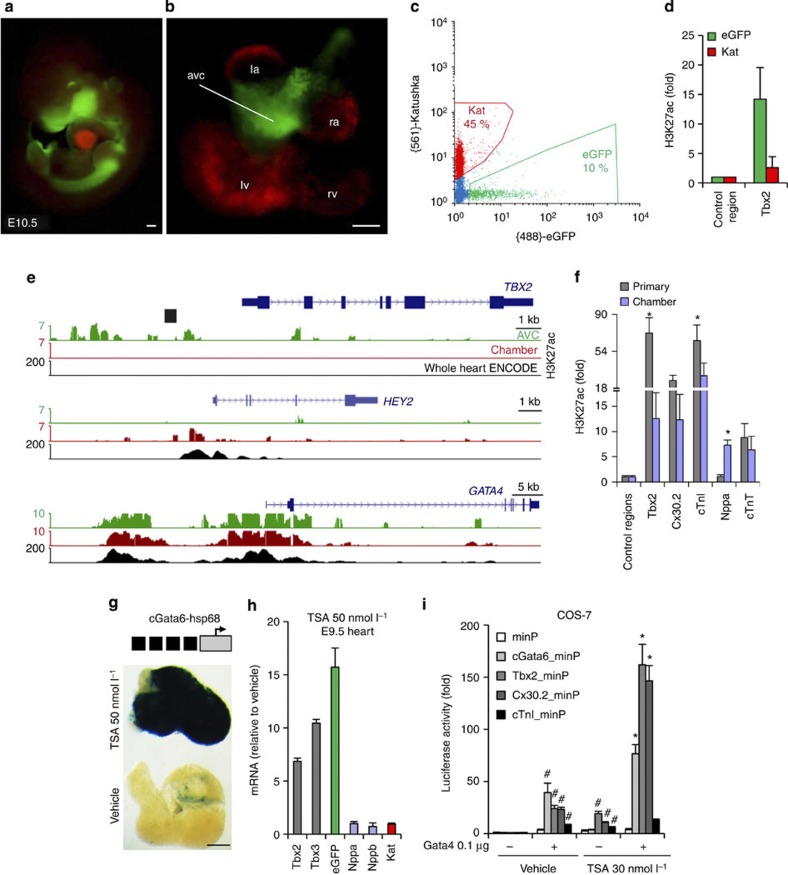
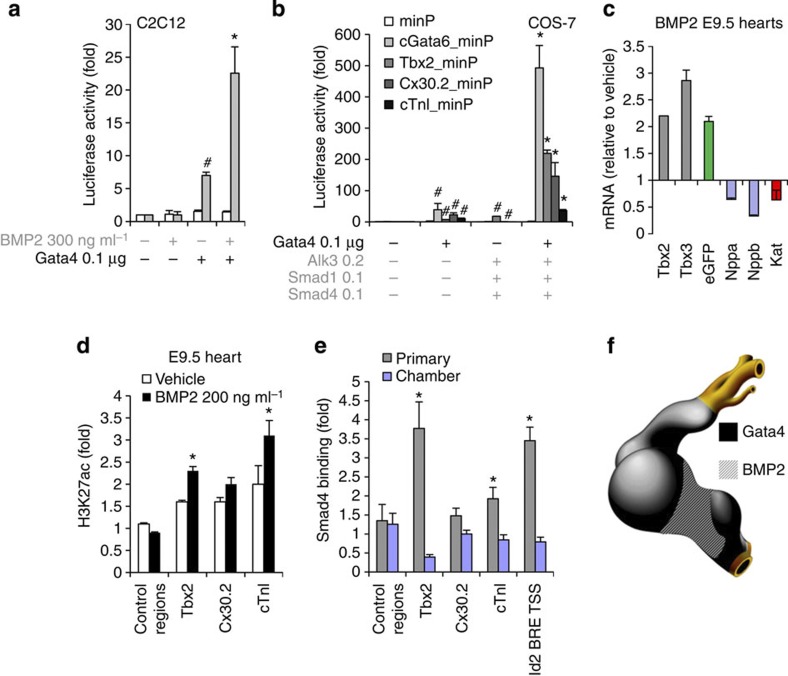
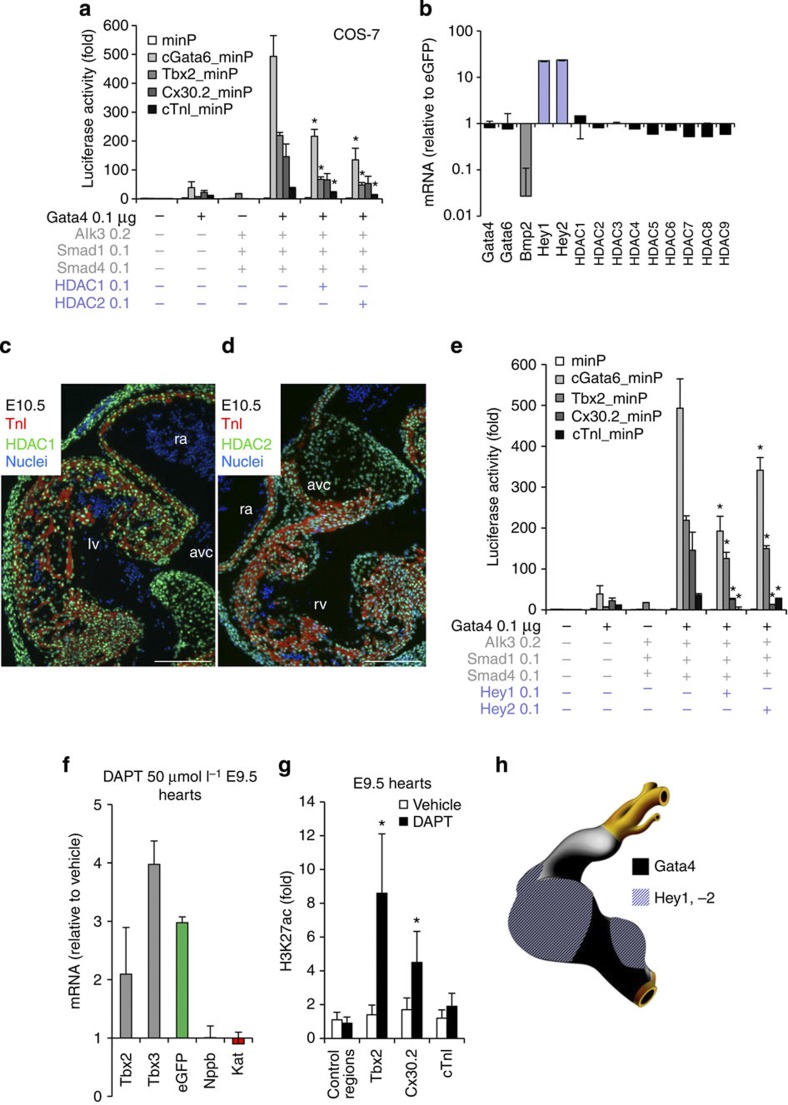
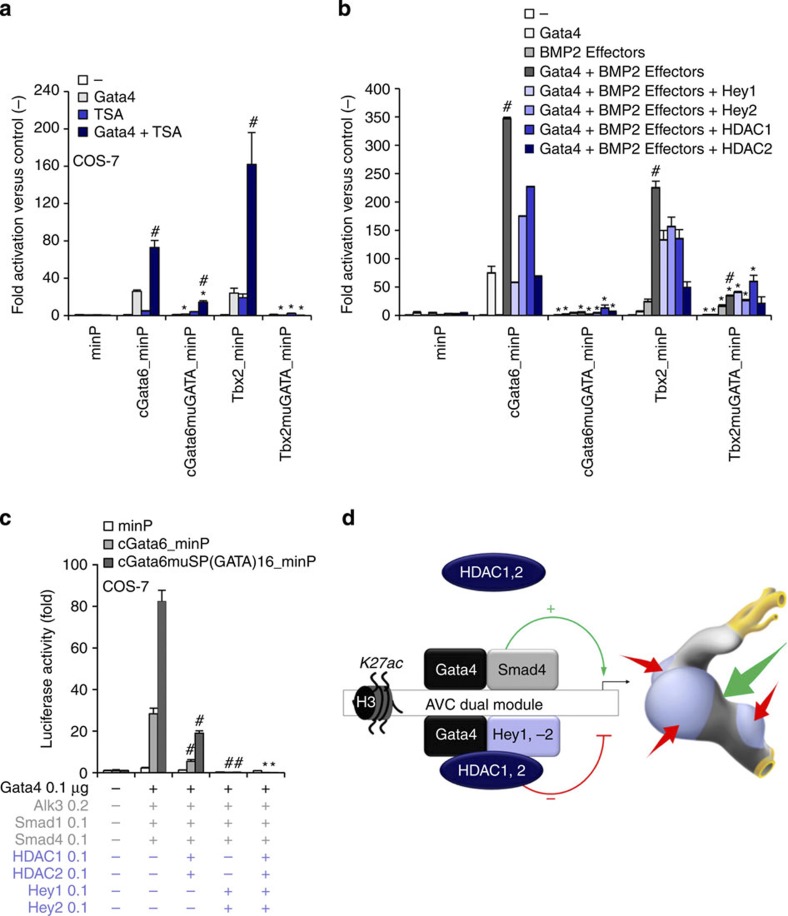
The embryonic vertebrate heart tube develops an atrioventricular canal that divides the atrial and ventricular chambers, forms atrioventricular conduction tissue and organizes valve development. Here we assess the transcriptional mechanism underlying this localized differentiation process. We show that atrioventricular canal-specific enhancers are GATA-binding site-dependent and act as switches that repress gene activity in the chambers. We find that atrioventricular canal-specific gene loci are enriched in H3K27ac, a marker of active enhancers, in atrioventricular canal tissue and depleted in H3K27ac in chamber tissue. In the atrioventricular canal, Gata4 activates the enhancers in synergy with Bmp2/Smad signalling, leading to H3K27 acetylation. In contrast, in chambers, Gata4 cooperates with pan-cardiac Hdac1 and Hdac2 and chamber-specific Hey1 and Hey2, leading to H3K27 deacetylation and repression. We conclude that atrioventricular canal-specific enhancers are platforms integrating cardiac transcription factors, broadly active histone modification enzymes and localized co-factors to drive atrioventricular canal-specific gene activity.
Figures
References
-
- Jensen B., Wang T., Christoffels V. M. & Moorman A. F. Evolution and development of the building plan of the vertebrate heart. Biochim. Biophys. Acta 1833, 783–794 (2013). - PubMed
-
- Hoffman J. I. & Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 39, 1890–1900 (2002). - PubMed
-
- Pierpont M. E., Markwald R. R. & Lin A. E. Genetic aspects of atrioventricular septal defects. Am. J. Med. Genet. 97, 289–296 (2000). - PubMed
-
- Walsh E. P. & Cecchin F. Arrhythmias in adult patients with congenital heart disease. Circulation 115, 534–545 (2007). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
