

Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis
- PMID: 24780756
- PMCID: PMC4079842
- DOI: 10.1158/0008-5472.CAN-13-3596
Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis
Abstract
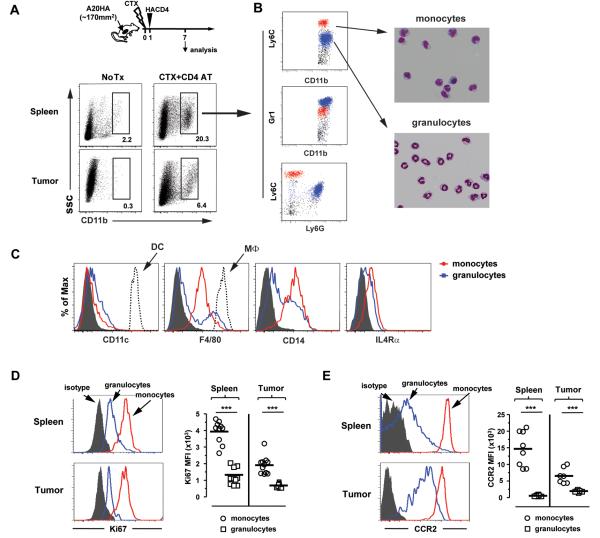
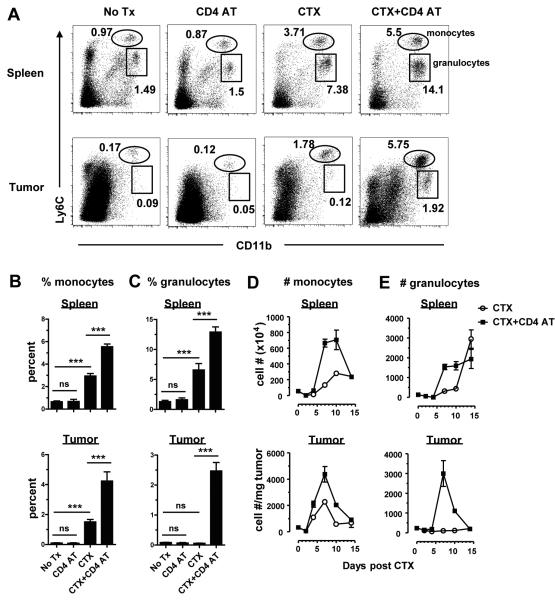
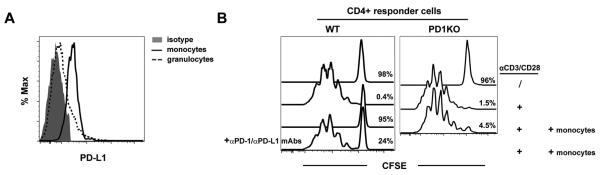
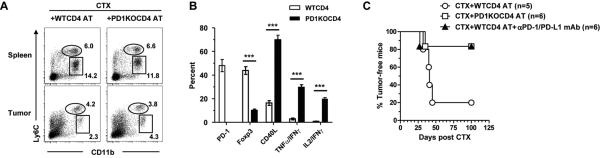
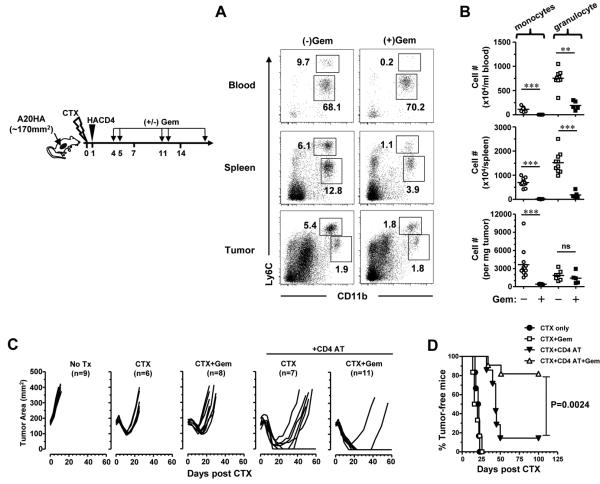
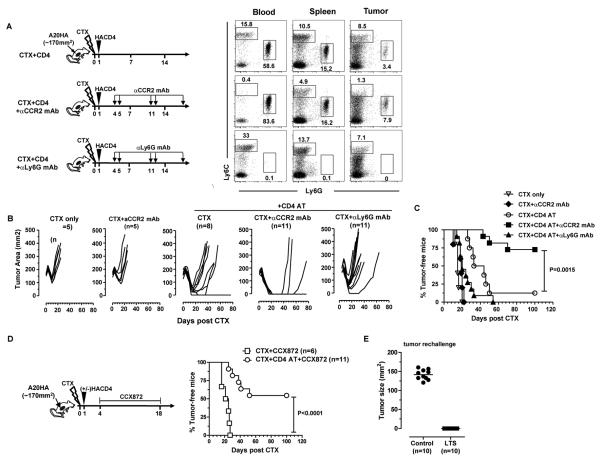
In recent years, immune-based therapies have become an increasingly attractive treatment option for patients with cancer. Cancer immunotherapy is often used in combination with conventional chemotherapy for synergistic effects. The alkylating agent cyclophosphamide (CTX) has been included in various chemoimmunotherapy regimens because of its well-known immunostimulatory effects. Paradoxically, cyclophosphamide can also induce suppressor cells that inhibit immune responses. However, the identity and biologic relevance of these suppressor cells are poorly defined. Here we report that cyclophosphamide treatment drives the expansion of inflammatory monocytic myeloid cells (CD11b(+)Ly6C(hi)CCR2(hi)) that possess immunosuppressive activities. In mice with advanced lymphoma, adoptive transfer (AT) of tumor-specific CD4(+) T cells following cyclophosphamide treatment (CTX+CD4 AT) provoked a robust initial antitumor immune response, but also resulted in enhanced expansion of monocytic myeloid cells. These therapy-induced monocytes inhibited long-term tumor control and allowed subsequent relapse by mediating functional tolerization of antitumor CD4(+) effector cells through the PD-1-PD-L1 axis. PD-1/PD-L1 blockade after CTX+CD4 AT therapy led to persistence of CD4(+) effector cells and durable antitumor effects. Depleting proliferative monocytes by administering low-dose gemcitabine effectively prevented tumor recurrence after CTX+CD4 AT therapy. Similarly, targeting inflammatory monocytes by disrupting the CCR2 signaling pathway markedly potentiated the efficacy of cyclophosphamide-based therapy. Besides cyclophosphamide, we found that melphalan and doxorubicin can also induce monocytic myeloid suppressor cells. These findings reveal a counter-regulation mechanism elicited by certain chemotherapeutic agents and highlight the importance of overcoming this barrier to prevent late tumor relapse after chemoimmunotherapy.
©2014 American Association for Cancer Research.
Figures

References
-
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. - PubMed
-
- Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. - PubMed
-
- Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011;33:369–383. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01HL56067/HL/NHLBI NIH HHS/United States
- P01AI056299/AI/NIAID NIH HHS/United States
- P01 AI056299/AI/NIAID NIH HHS/United States
- U01 AI083005/AI/NIAID NIH HHS/United States
- R01 CA133085/CA/NCI NIH HHS/United States
- R01CA158202/CA/NCI NIH HHS/United States
- R01 CA103320/CA/NCI NIH HHS/United States
- R01 AI034495/AI/NIAID NIH HHS/United States
- R01CA112431/CA/NCI NIH HHS/United States
- R01 CA158202/CA/NCI NIH HHS/United States
- AI103347/AI/NIAID NIH HHS/United States
- R01 HL056067/HL/NHLBI NIH HHS/United States
- R01 CA072669/CA/NCI NIH HHS/United States
- R01 CA112431/CA/NCI NIH HHS/United States
- R01 AI103347/AI/NIAID NIH HHS/United States
- AI083005/AI/NIAID NIH HHS/United States
- P01 CA065493/CA/NCI NIH HHS/United States
- R01 CA096651/CA/NCI NIH HHS/United States
- R01CA72669/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
