

Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes
- PMID: 24782769
- PMCID: PMC3986552
- DOI: 10.3389/fphar.2014.00061
Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes
Abstract
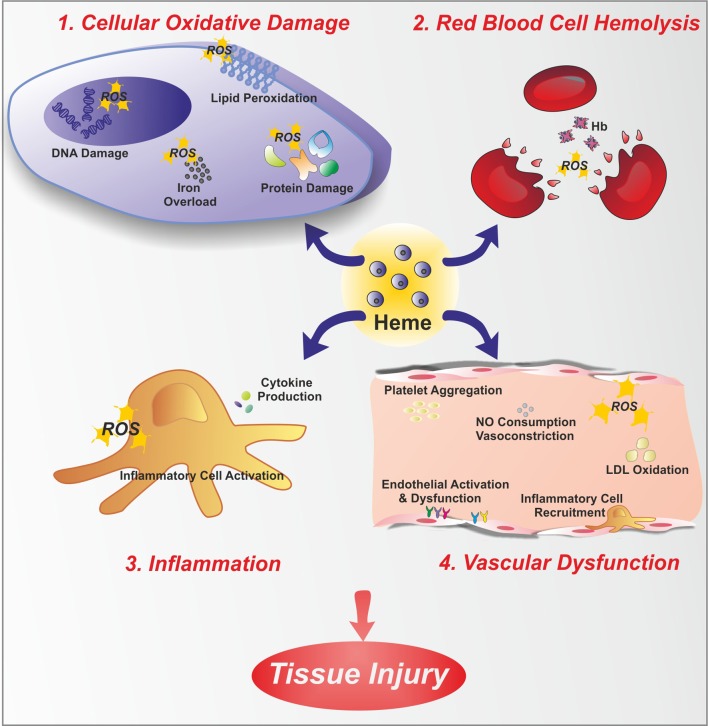
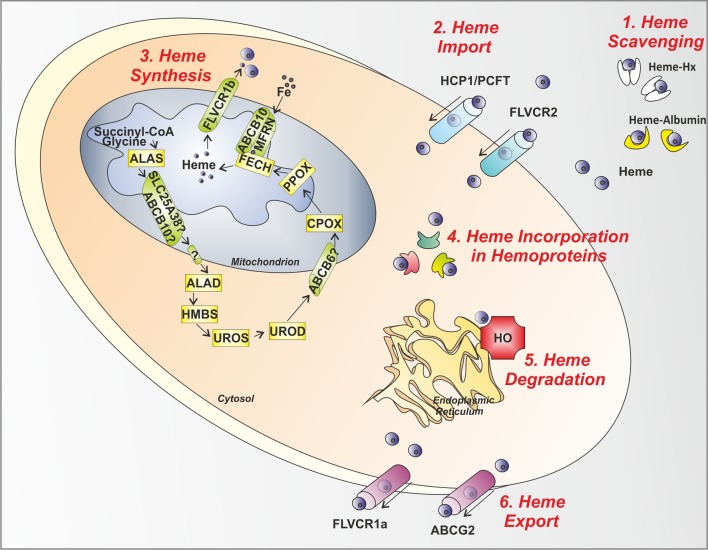
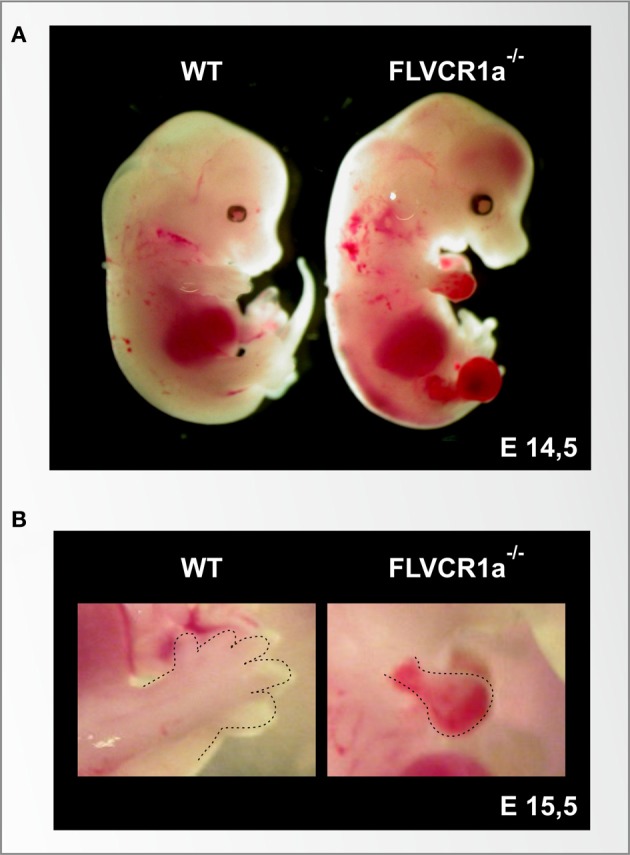
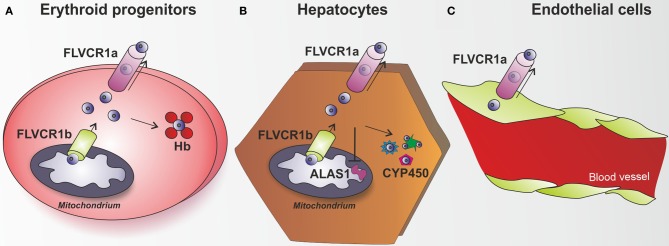
Heme (iron-protoporphyrin IX) is an essential co-factor involved in multiple biological processes: oxygen transport and storage, electron transfer, drug and steroid metabolism, signal transduction, and micro RNA processing. However, excess free-heme is highly toxic due to its ability to promote oxidative stress and lipid peroxidation, thus leading to membrane injury and, ultimately, apoptosis. Thus, heme metabolism needs to be finely regulated. Intracellular heme amount is controlled at multiple levels: synthesis, utilization by hemoproteins, degradation and both intracellular and intercellular trafficking. This review focuses on recent findings highlighting the importance of controlling intracellular heme levels to counteract heme-induced oxidative stress. The contributions of heme scavenging from the extracellular environment, heme synthesis and incorporation into hemoproteins, heme catabolism and heme transport in maintaining adequate intracellular heme content are discussed. Particular attention is put on the recently described mechanisms of heme trafficking through the plasma membrane mediated by specific heme importers and exporters. Finally, the involvement of genes orchestrating heme metabolism in several pathological conditions is illustrated and new therapeutic approaches aimed at controlling heme metabolism are discussed.
Keywords: ABCG2; FLVCR1; FLVCR2; HCP1/PCFT; HO-1; hemopexin.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases