

Presynaptic inhibition of spinal sensory feedback ensures smooth movement
- PMID: 24784215
- PMCID: PMC4292914
- DOI: 10.1038/nature13276
Presynaptic inhibition of spinal sensory feedback ensures smooth movement
Abstract
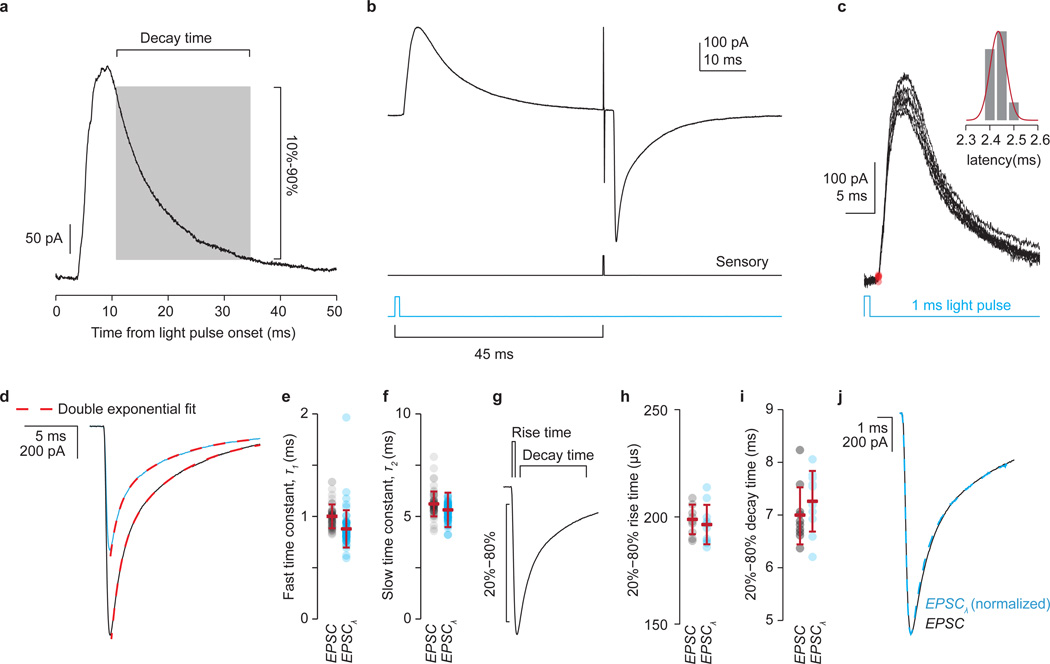
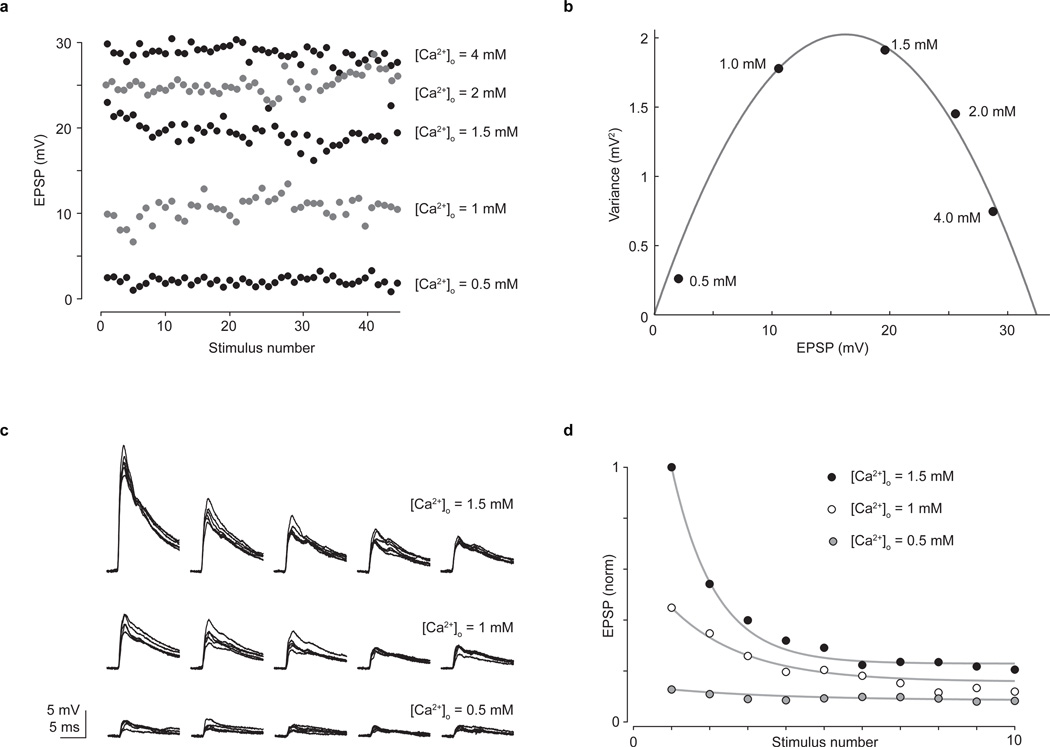
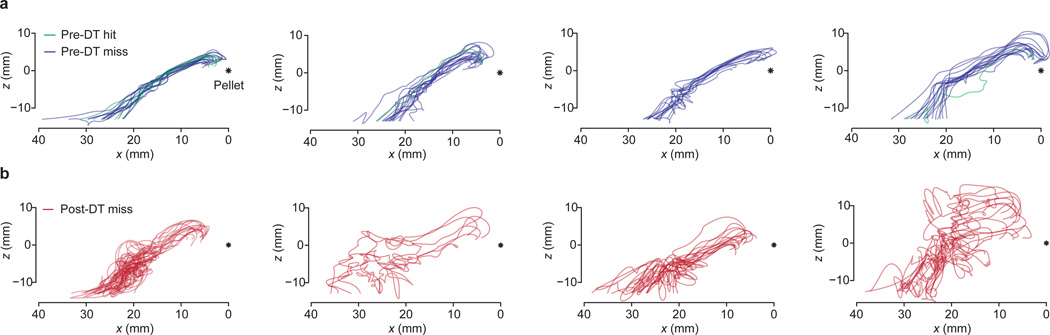
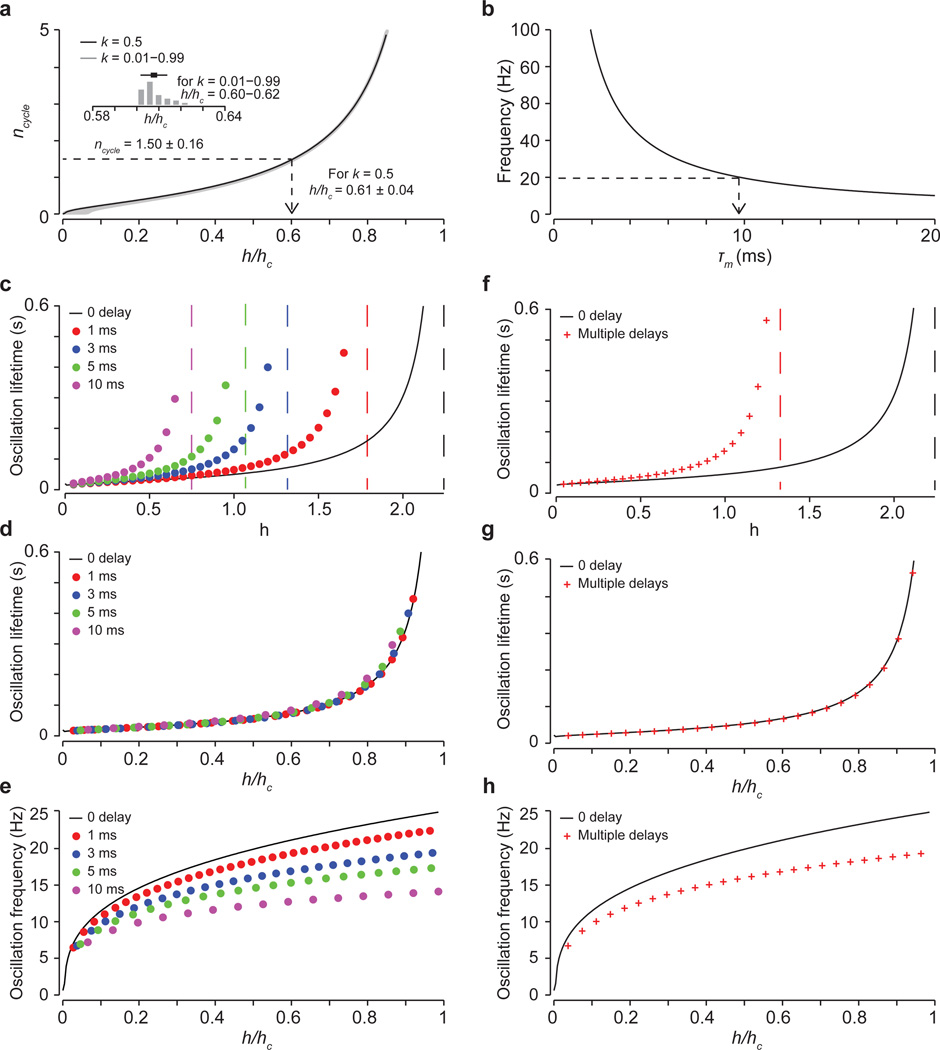
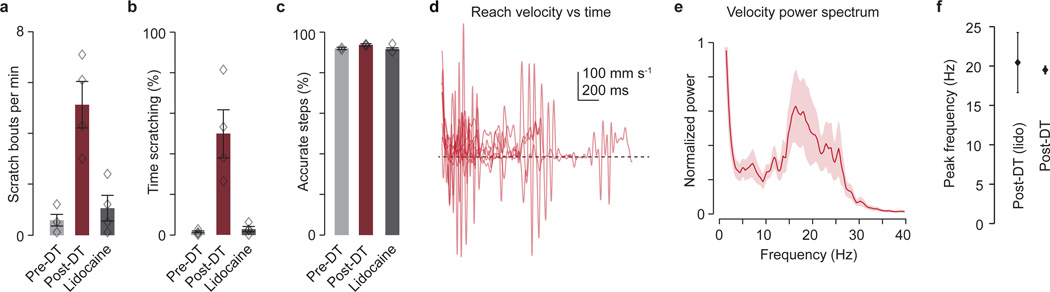
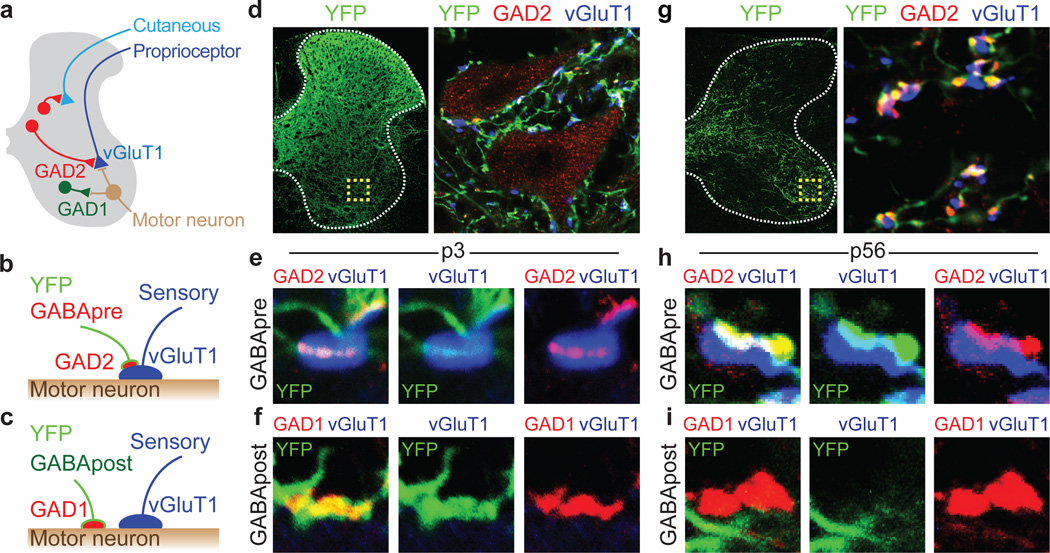
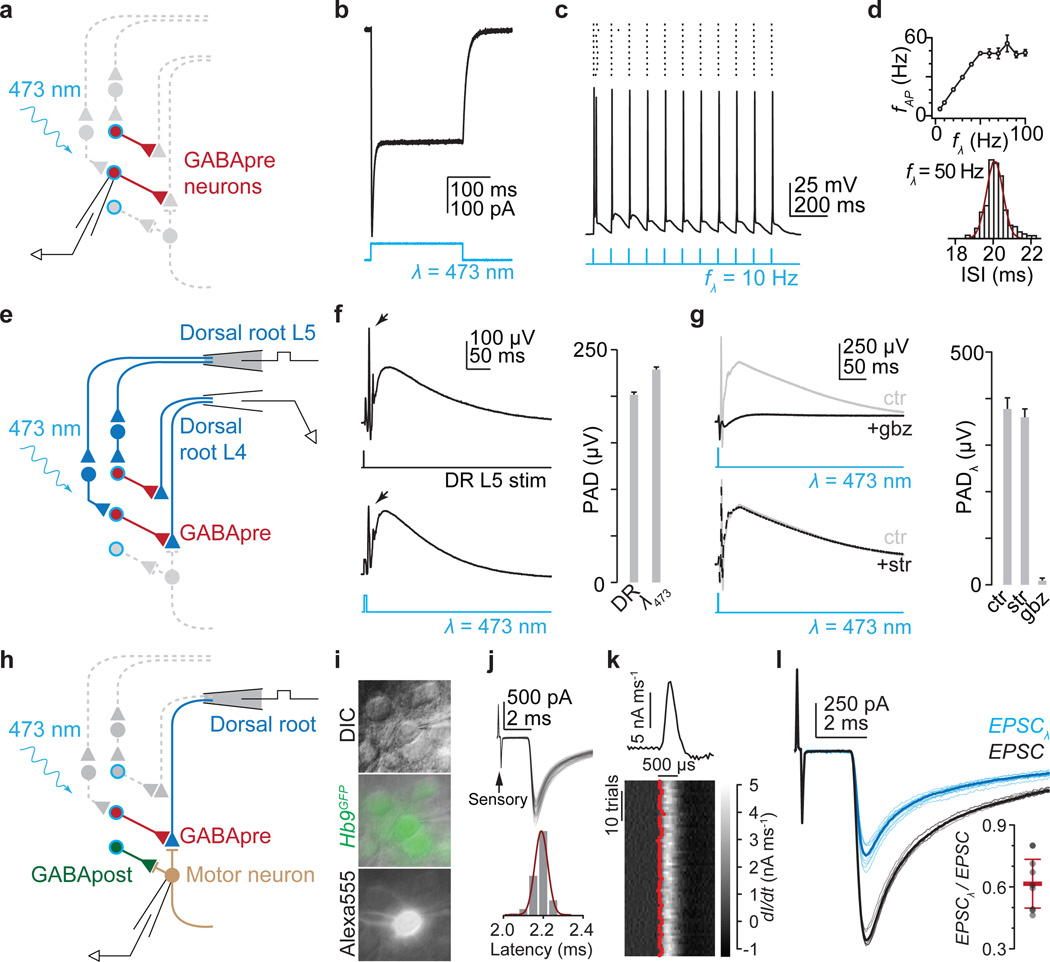
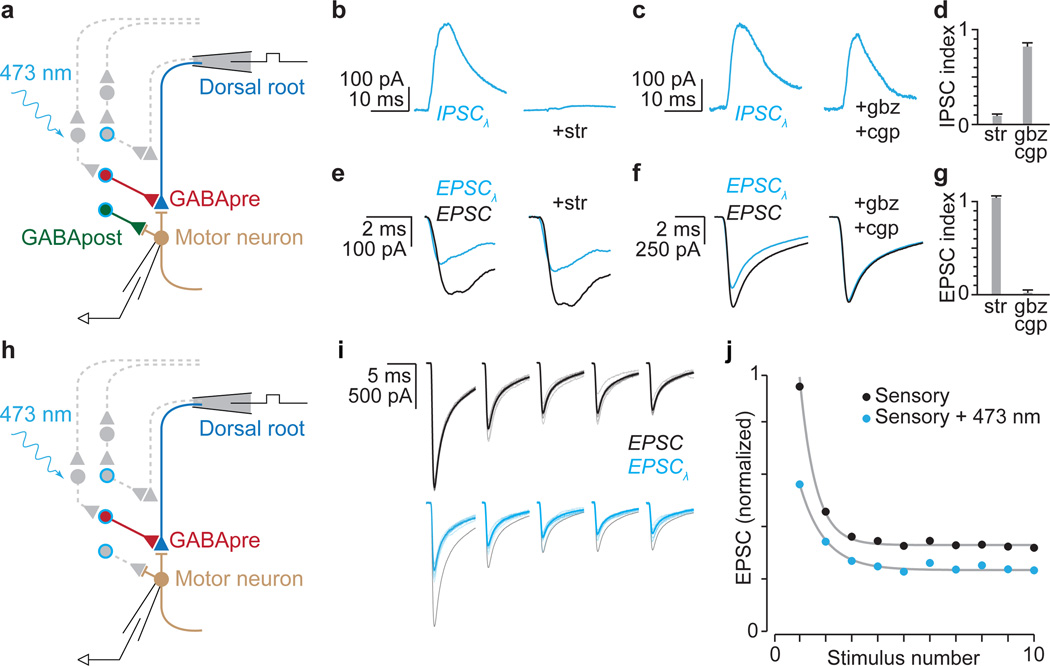
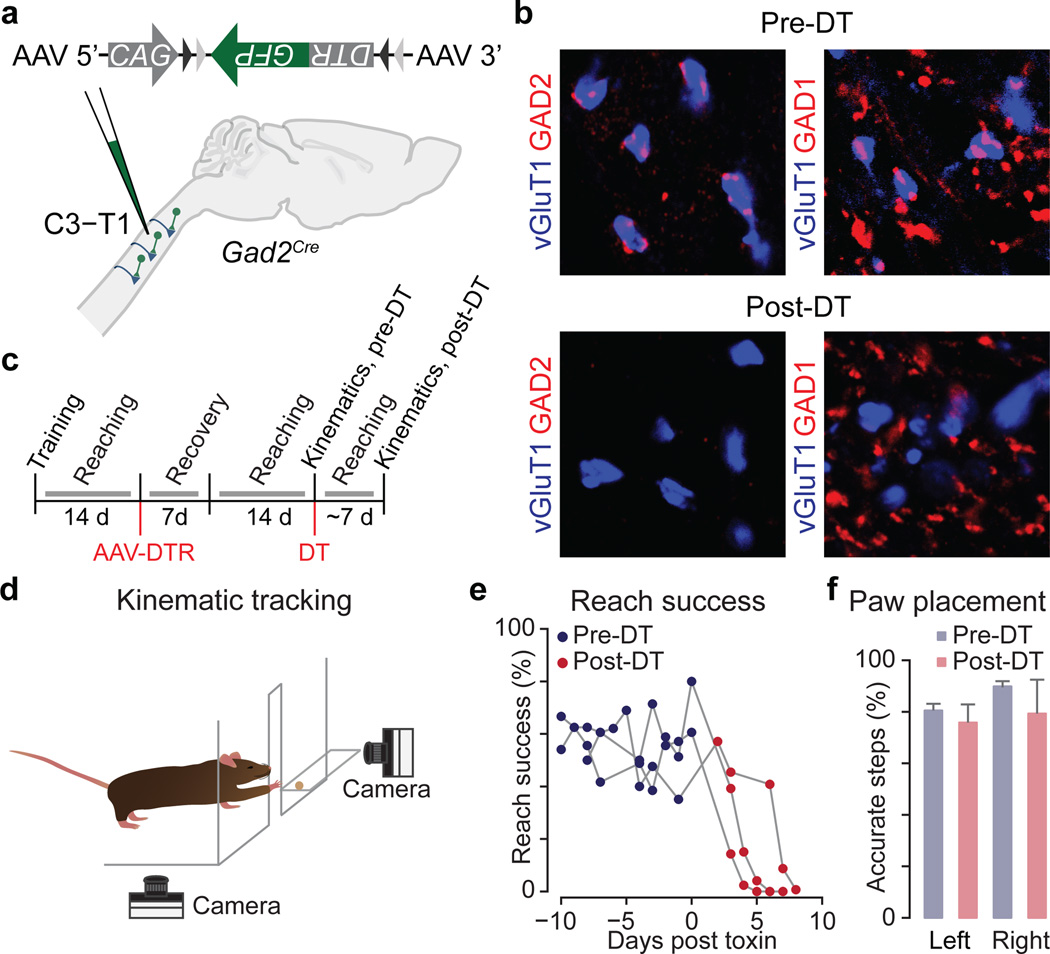
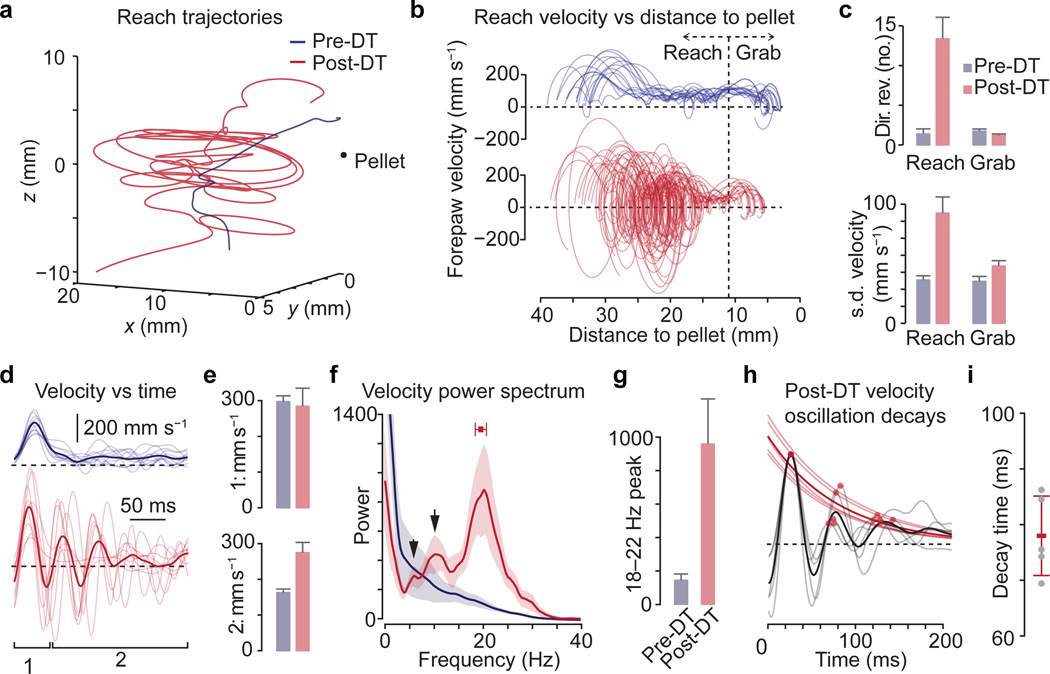
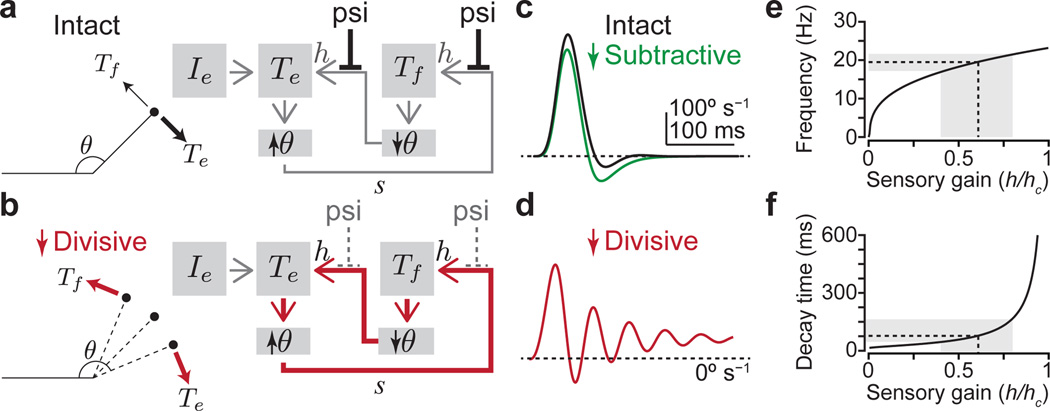
The precision of skilled movement depends on sensory feedback and its refinement by local inhibitory microcircuits. One specialized set of spinal GABAergic interneurons forms axo-axonic contacts with the central terminals of sensory afferents, exerting presynaptic inhibitory control over sensory-motor transmission. The inability to achieve selective access to the GABAergic neurons responsible for this unorthodox inhibitory mechanism has left unresolved the contribution of presynaptic inhibition to motor behaviour. We used Gad2 as a genetic entry point to manipulate the interneurons that contact sensory terminals, and show that activation of these interneurons in mice elicits the defining physiological characteristics of presynaptic inhibition. Selective genetic ablation of Gad2-expressing interneurons severely perturbs goal-directed reaching movements, uncovering a pronounced and stereotypic forelimb motor oscillation, the core features of which are captured by modelling the consequences of sensory feedback at high gain. Our findings define the neural substrate of a genetically hardwired gain control system crucial for the smooth execution of movement.
Figures
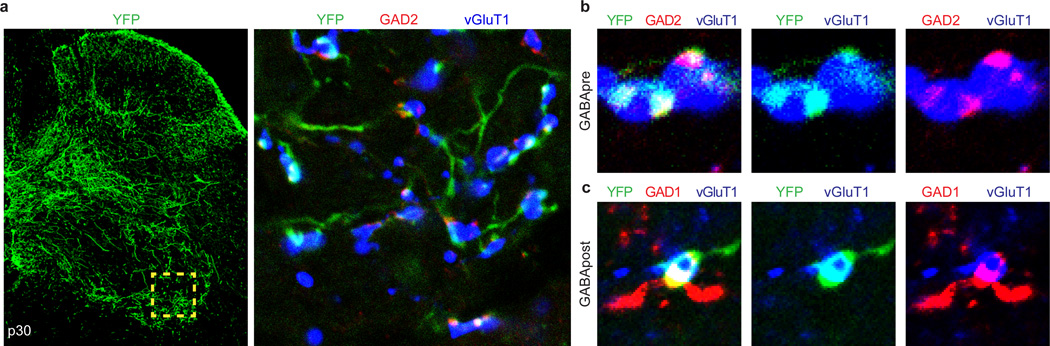
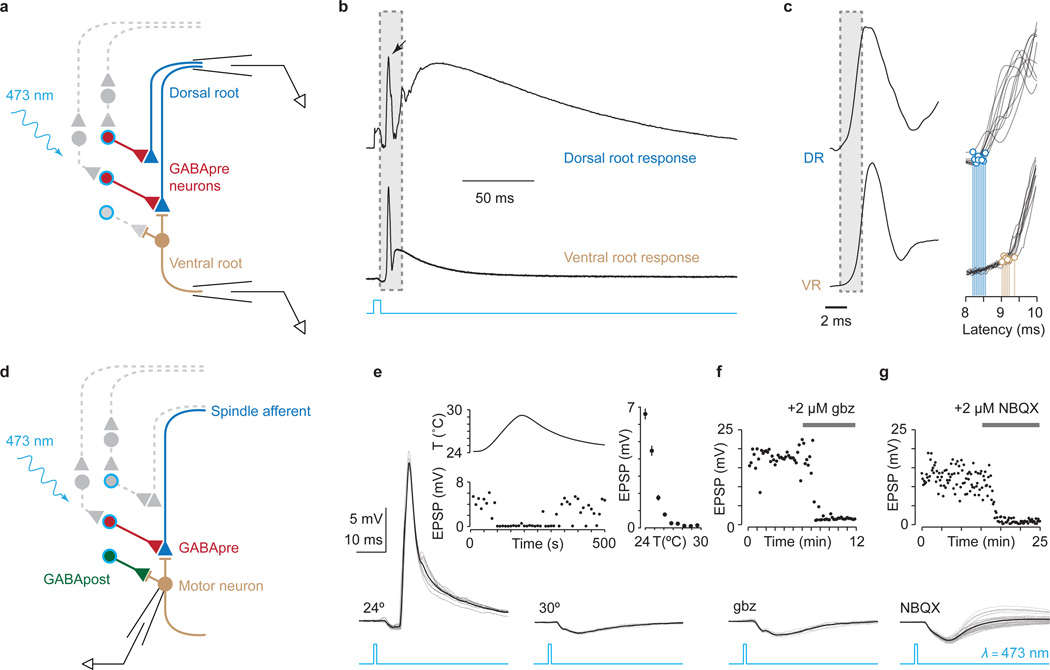
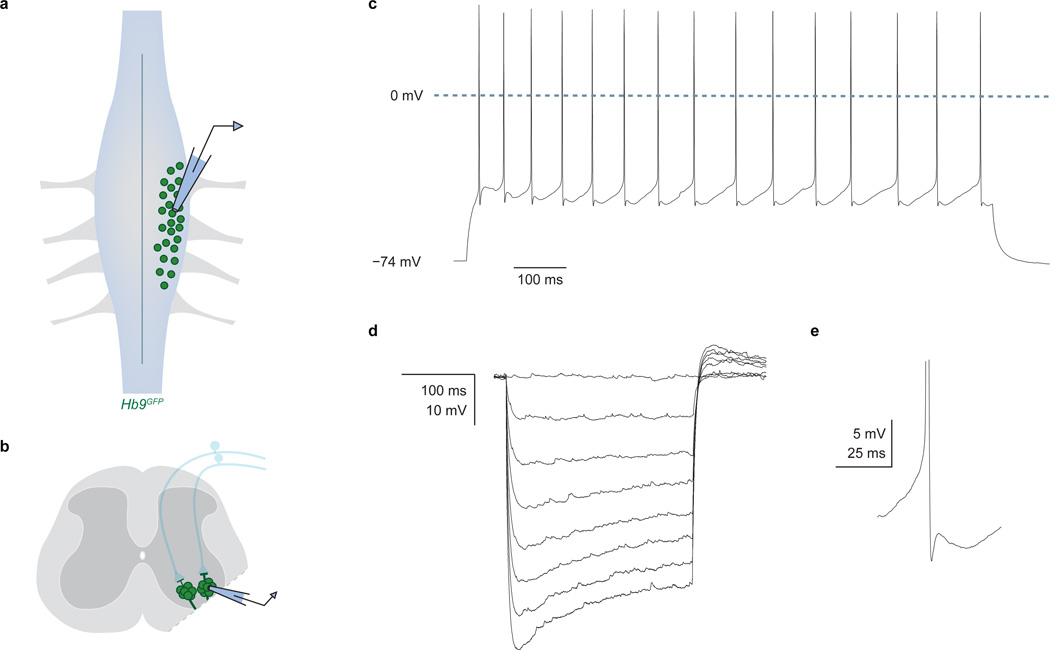
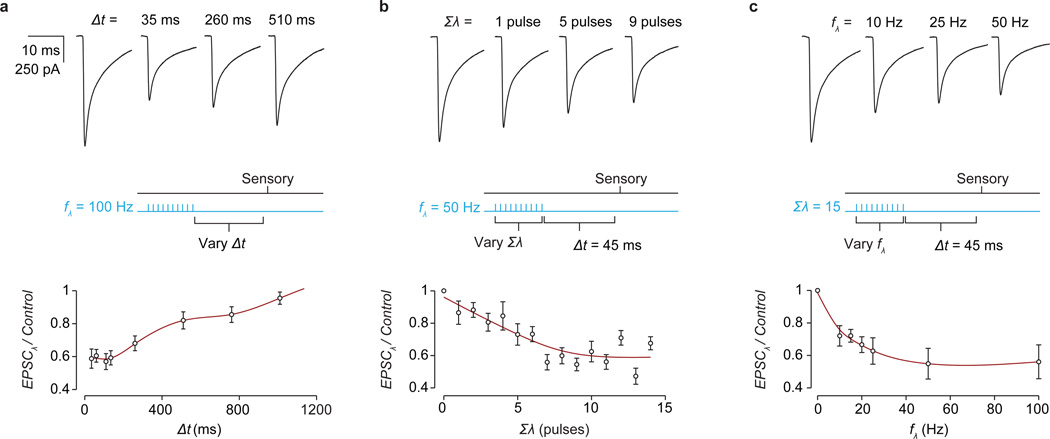
Comment in
-
Neuroscience: Feedback throttled down for smooth moves.Nature. 2014 May 1;509(7498):38-9. doi: 10.1038/509038a. Nature. 2014. PMID: 24784211 No abstract available.
References
-
- Windhorst U. Muscle proprioceptive feedback and spinal networks. Brain Research Bulletin. 2007;73:155–202. - PubMed
-
- Arber S. Motor circuits in action: specification, connectivity, and function. Neuron. 2012;74:975–989. - PubMed
-
- Frank K, Fuortes M. Presynaptic and postsynaptic inhibition of monosynaptic reflexes. Federation Proceedings. 1957;16:49–50.
-
- Lundberg A. Supraspinal control of transmission in reflex paths to motoneurones and primary afferents. Progress in Brain Research. 1964;12:197–221. - PubMed
Supplementary References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
