

Trauma-Induced Coagulopathy: An Institution's 35 Year Perspective on Practice and Research
- PMID: 24786172
- PMCID: PMC4214916
- DOI: 10.1177/1457496914531927
Trauma-Induced Coagulopathy: An Institution's 35 Year Perspective on Practice and Research
Abstract
Introduction: Injury is the second leading cause of death worldwide, and as much as 40% of injury-related mortality is attributed to uncontrollable hemorrhage. This persists despite establishment of regionalized trauma systems and advances in the management of severely injured patients. Trauma-induced coagulopathy has been identified as the most common preventable cause of postinjury mortality.
Methods: A review of the current literature was performed by collecting PUBMED references related to trauma-induced coagulopathy. Data were then critically analyzed and summarized based on the authors' clinical and research perspective, as well as that reported by other institutions and researchers interested in trauma-induced coagulopathy. A particular focus was placed on those aspects of coagulopathy in which agreement among clinical and basic scientists is currently lacking; these include, pathophysiology, the role of blood components and factor therapy, and goal-directed assessment and management.
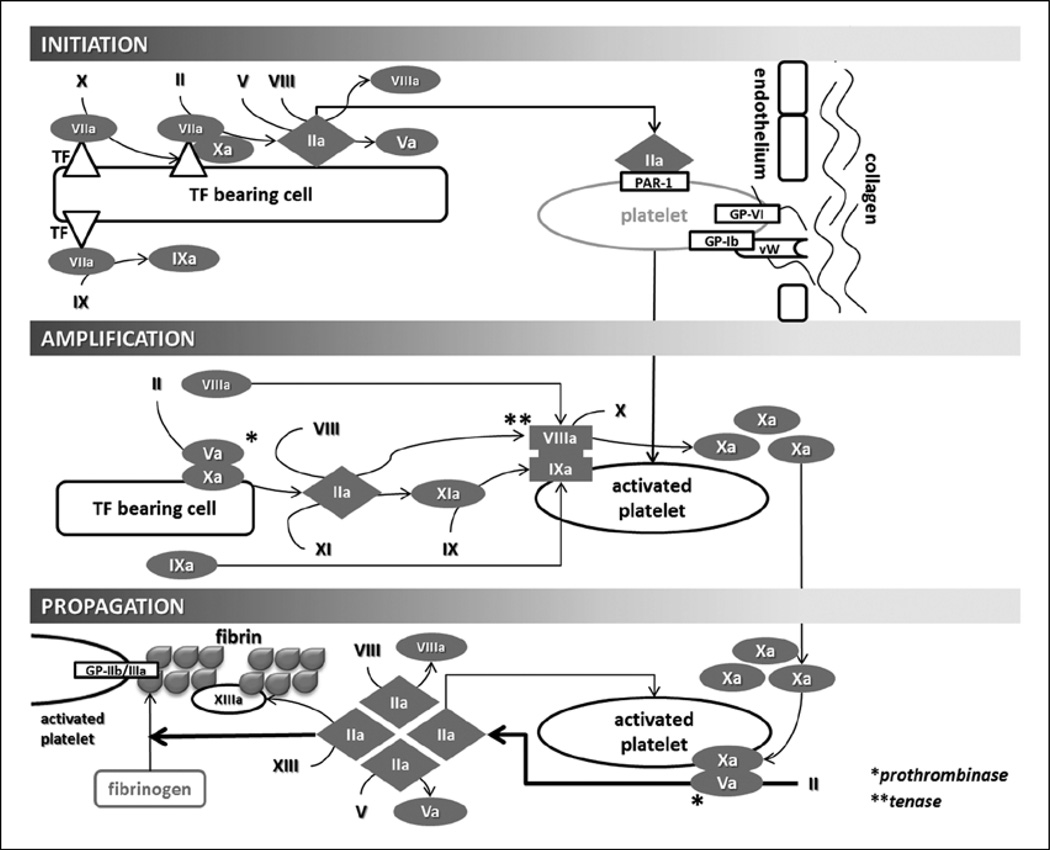
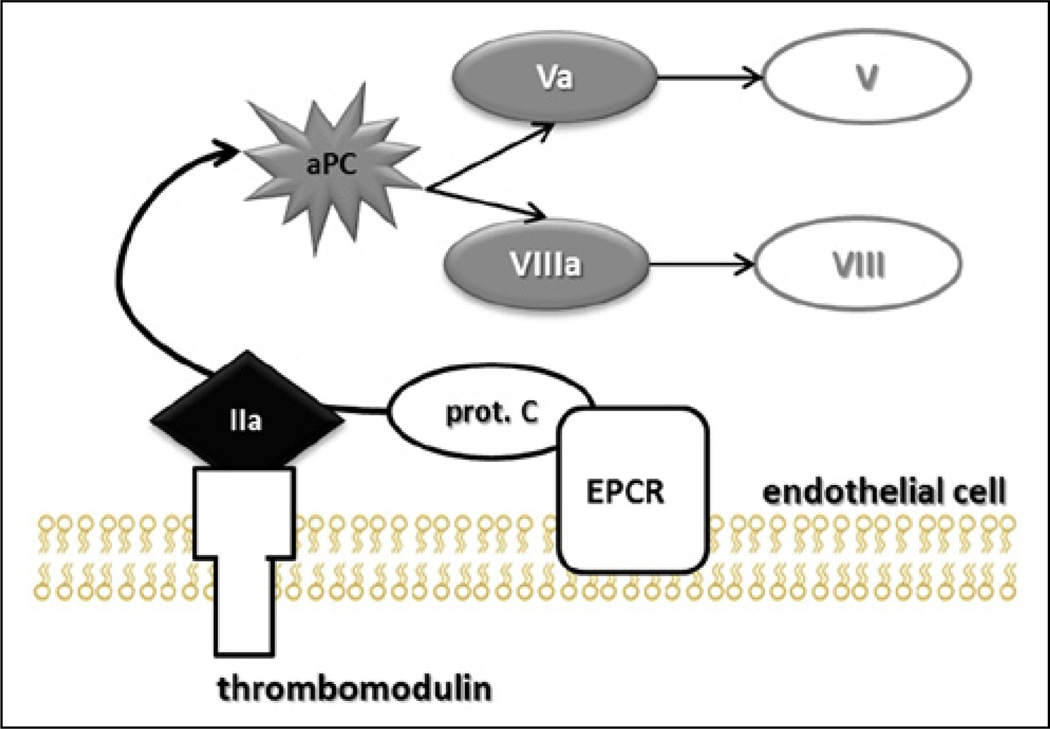
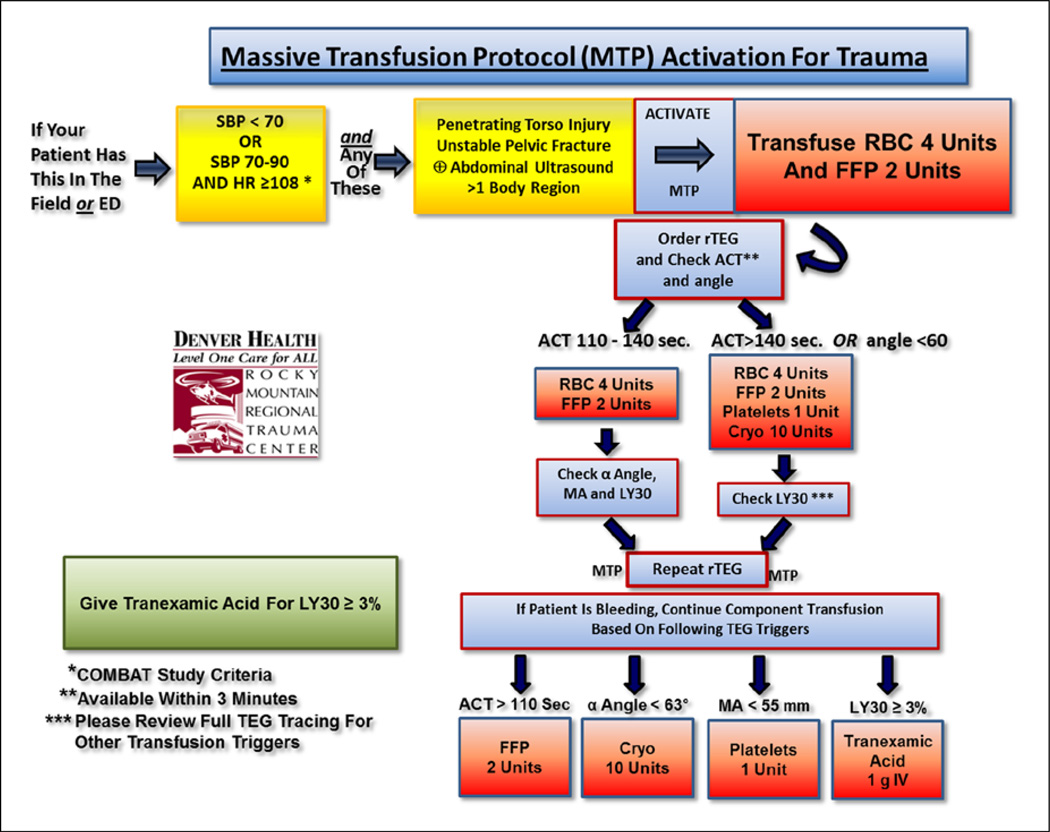
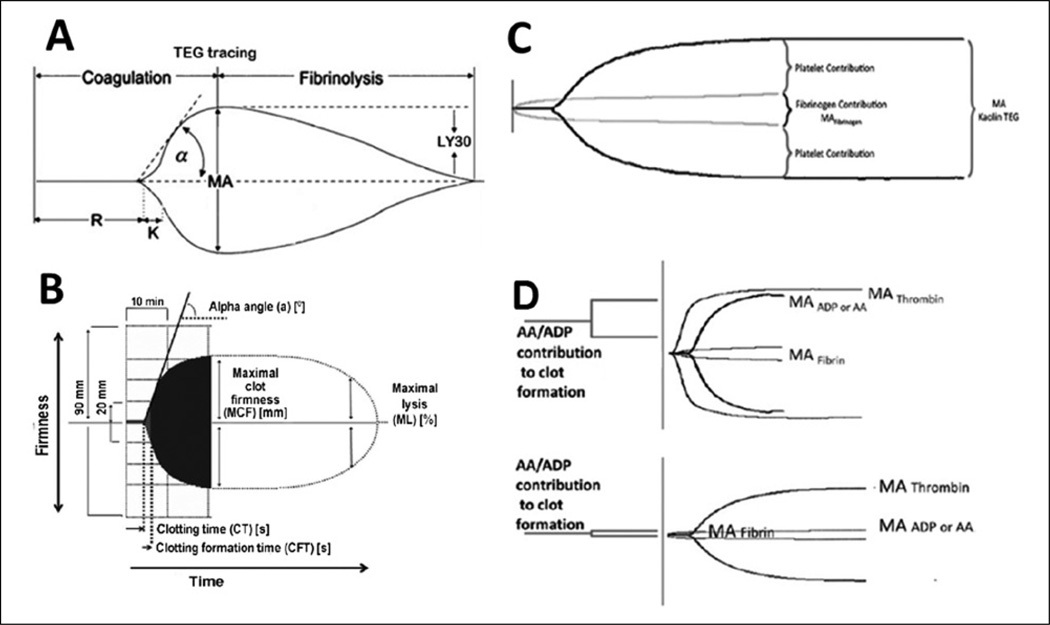
Results: Trauma-induced coagulopathy has been recognized in approximately one-third of trauma patients. There is a vast range of severity, and the emergence of viscoelastic assays, such as thrombelastography and rotational thromboelastogram, has refined its diagnosis and management, particularly through the establishment of goal-directed massive transfusion protocols. Despite advancements in the diagnosis and management of trauma-induced coagulopathy, much remains to be understood regarding its pathophysiology. The cell-based model of hemostasis has allowed for characterization of endothelial dysfunction, impaired thrombin generation, platelet dysfunction, fibrinolysis, endogenous anticoagulants such as protein-C, and antifibrinolytic proteins. These concepts collectively compose the contemporary, but still partial, understanding of trauma-induced coagulopathy.
Conclusion: Trauma-induced coagulopathy is a complex pathophysiological condition, of which some mechanisms have been characterized, but much remains to be understood in order to translate this knowledge into improved outcomes for the injured patient.
Keywords: Coagulopathy; ROTEM; fibrinolysis; hemorrhage; thrombelastography; transfusion; trauma.
© The Finnish Surgical Society 2014.
Conflict of interest statement
DECLARATION OF CONFLICTING INTERESTS None.
Figures


References
-
- Sauaia A, Moore FA, Moore EE, et al. Epidemiology of trauma deaths: A reassessment. J Trauma. 1995;38:185–193. - PubMed
-
- Scott R, Jr, Crosby WH. Changes in the coagulation mechanism following wounding and resuscitation with stored blood; A study of battle casualties in Korea. Blood. 1954;9:609–621. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
