

Whole-genome-based Mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach
- PMID: 24789177
- PMCID: PMC4097744
- DOI: 10.1128/JCM.00567-14
Whole-genome-based Mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach
Abstract
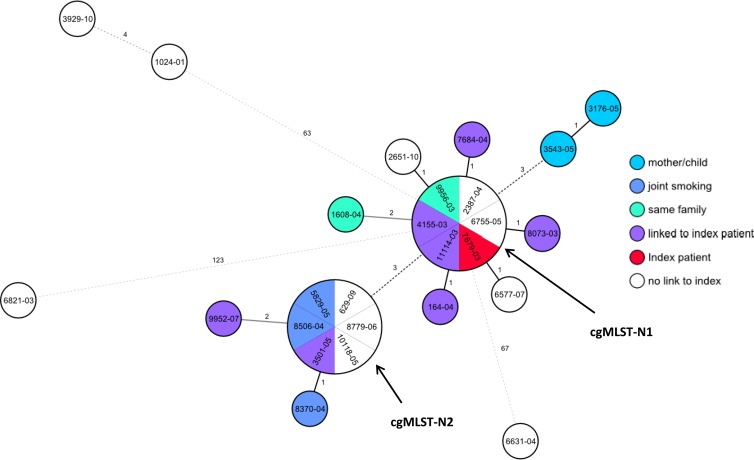
Whole-genome sequencing (WGS) allows for effective tracing of Mycobacterium tuberculosis complex (MTBC) (tuberculosis pathogens) transmission. However, it is difficult to standardize and, therefore, is not yet employed for interlaboratory prospective surveillance. To allow its widespread application, solutions for data standardization and storage in an easily expandable database are urgently needed. To address this question, we developed a core genome multilocus sequence typing (cgMLST) scheme for clinical MTBC isolates using the Ridom SeqSphere(+) software, which transfers the genome-wide single nucleotide polymorphism (SNP) diversity into an allele numbering system that is standardized, portable, and not computationally intensive. To test its performance, we performed WGS analysis of 26 isolates with identical IS6110 DNA fingerprints and spoligotyping patterns from a longitudinal outbreak in the federal state of Hamburg, Germany (notified between 2001 and 2010). The cgMLST approach (3,041 genes) discriminated the 26 strains with a resolution comparable to that of SNP-based WGS typing (one major cluster of 22 identical or closely related and four outlier isolates with at least 97 distinct SNPs or 63 allelic variants). Resulting tree topologies are highly congruent and grouped the isolates in both cases analogously. Our data show that SNP- and cgMLST-based WGS analyses facilitate high-resolution discrimination of longitudinal MTBC outbreaks. cgMLST allows for a meaningful epidemiological interpretation of the WGS genotyping data. It enables standardized WGS genotyping for epidemiological investigations, e.g., on the regional public health office level, and the creation of web-accessible databases for global TB surveillance with an integrated early warning system.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures




References
-
- Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J, Wirth T, Jaenicke S, Schuback S, Rüsch-Gerdes S, Supply P, Kalinowski J, Niemann S. 2013. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 10:e1001387. 10.1371/journal.pmed.1001387 - DOI - PMC - PubMed
-
- Bryant JM, Schürch AC, van Deutekom H, Harris SR, de Beer JL, de Jager V, Kremer K, van Hijum SAFT, Siezen RJ, Borgdorff M, Bentley SD, Parkhill J, van Soolingen D. 2013. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect. Dis. 13:110. 10.1186/1471-2334-13-110 - DOI - PMC - PubMed
-
- Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, Varhol R, Birol I, Lem M, Sharma MK, Elwood K, Jones SJM, Brinkman FSL, Brunham RC, Tang P. 2011. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 364:730–739. 10.1056/NEJMoa1003176 - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
