

Coordinated regulation of nuclear receptor CAR by CCRP/DNAJC7, HSP70 and the ubiquitin-proteasome system
- PMID: 24789201
- PMCID: PMC4008524
- DOI: 10.1371/journal.pone.0096092
Coordinated regulation of nuclear receptor CAR by CCRP/DNAJC7, HSP70 and the ubiquitin-proteasome system
Abstract
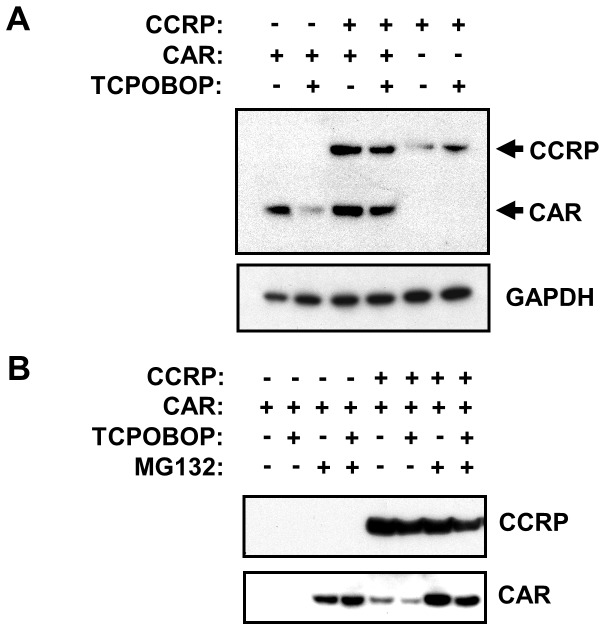
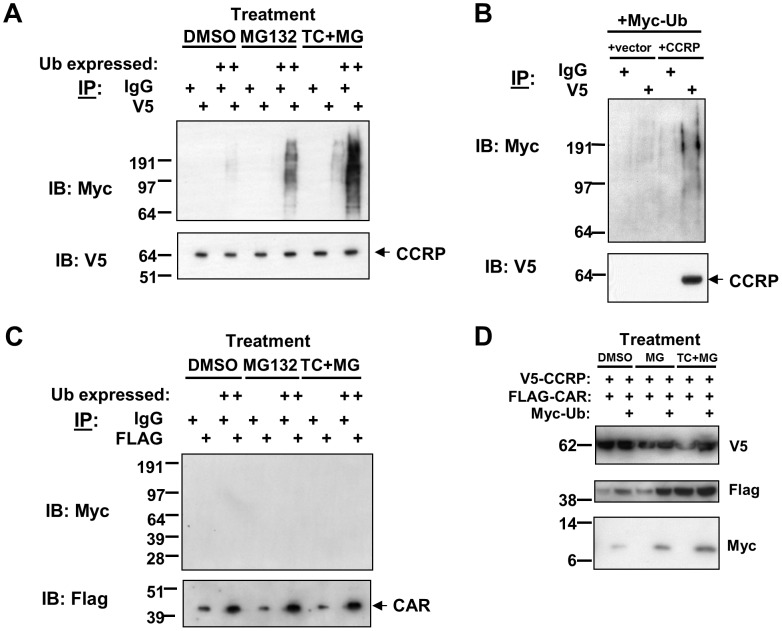
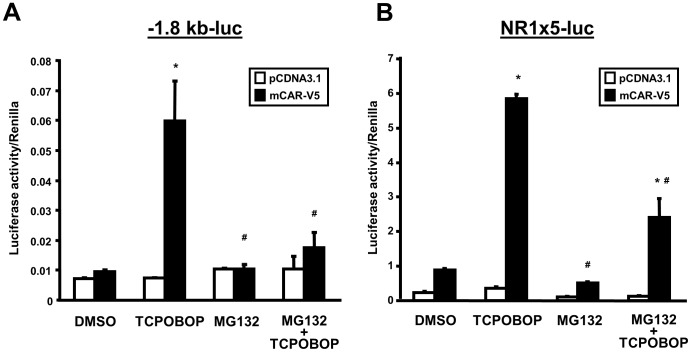
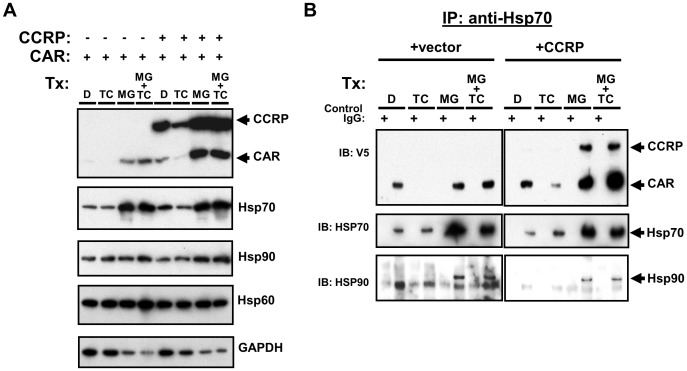
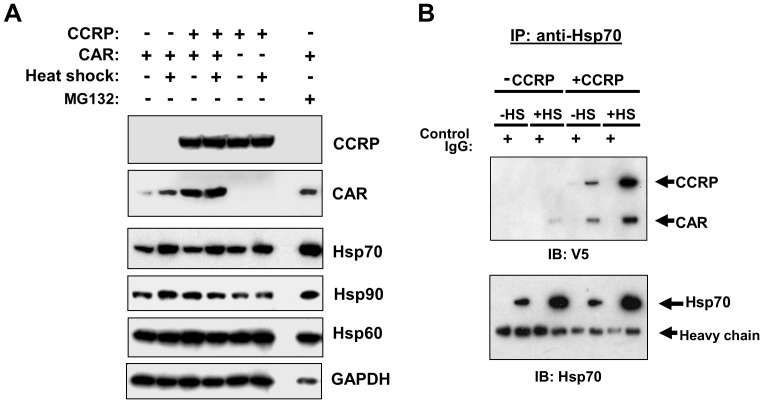
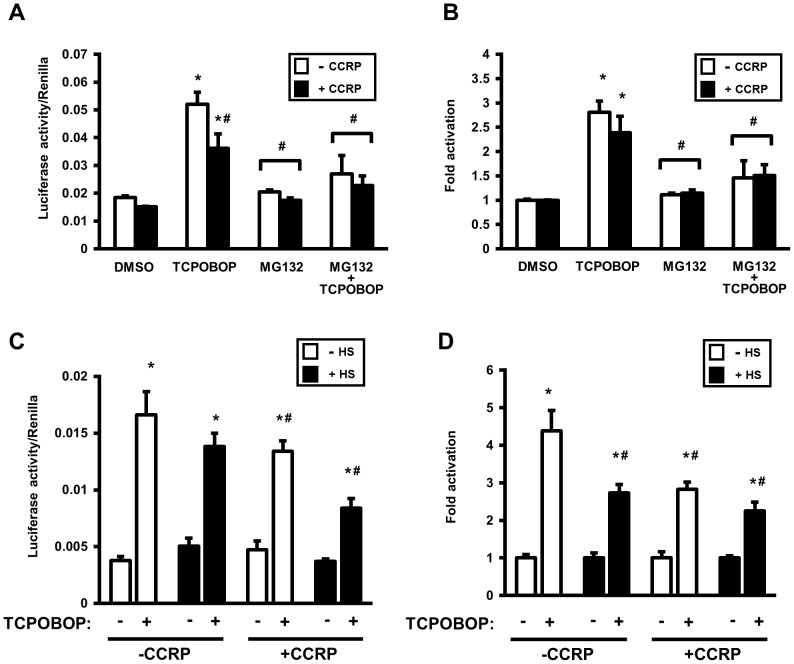
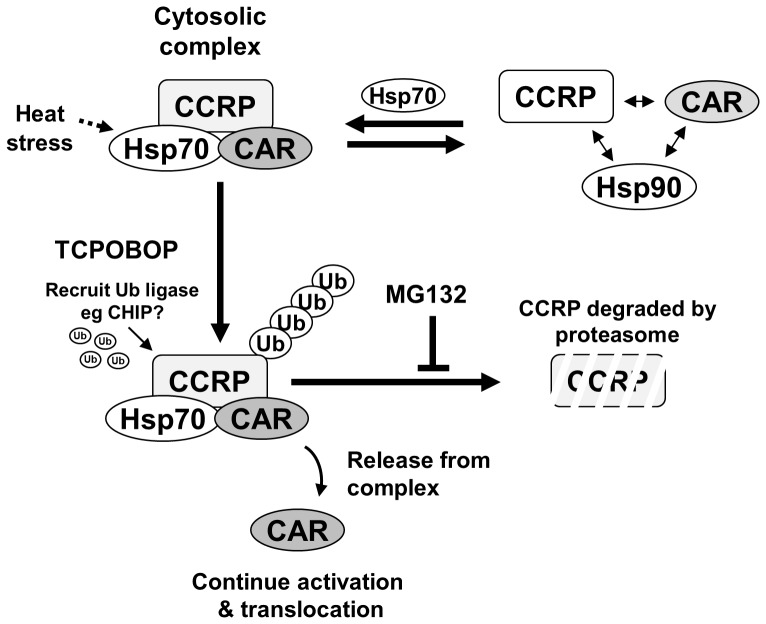
The constitutive active/androstane receptor (CAR) plays an important role as a coordinate transcription factor in the regulation of various hepatic metabolic pathways for chemicals such as drugs, glucose, fatty acids, bilirubin, and bile acids. Currently, it is known that in its inactive state, CAR is retained in the cytoplasm in a protein complex with HSP90 and the tetratricopeptide repeat protein cytosoplasmic CAR retention protein (CCRP). Upon activation by phenobarbital (PB) or the PB-like inducer 1,4-bis[2-(3,5-dichloropyridyloxy)]-benzene (TCPOBOP), CAR translocates into the nucleus. We have identified two new components to the cytoplasmic regulation of CAR: ubiquitin-dependent degradation of CCRP and protein-protein interaction with HSP70. Treatment with the proteasome inhibitor MG132 (5 µM) causes CAR to accumulate in the cytoplasm of transfected HepG2 cells. In the presence of MG132, TCPOBOP increases CCRP ubiquitination in HepG2 cells co-expressing CAR, while CAR ubiquitination was not detected. MG132 treatment of HepG2 also attenuated of TCPOBOP-induced CAR transcriptional activation on reporter constructs which contain CAR-binding DNA elements derived from the human CYP2B6 gene. The elevation of cytoplasmic CAR protein with MG132 correlated with an increase of HSP70, and to a lesser extent HSP60. Both CCRP and CAR were found to interact with endogenous HSP70 in HepG2 cells by immunoprecipitation analysis. Induction of HSP70 levels by heat shock also increased cytoplasmic CAR levels, similar to the effect of MG132. Lastly, heat shock attenuated TCPOBOP-induced CAR transcriptional activation, also similar to the effect of MG132. Collectively, these data suggest that ubiquitin-proteasomal regulation of CCRP and HSP70 are important contributors to the regulation of cytoplasmic CAR levels, and hence the ability of CAR to respond to PB or PB-like inducers.
Conflict of interest statement
Figures
References
-
- Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, et al. (2002) Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol 61: 1–6. - PubMed
-
- Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD (2000) The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature 407: 920–923. - PubMed
-
- Honkakoski P, Moore R, Gynther J, Negishi M (1996) Characterization of phenobarbital-inducible mouse Cyp2b10 gene transcription in primary hepatocytes. J Biol Chem 271: 9746–9753. - PubMed
-
- Honkakoski P, Negishi M (1998) Regulatory DNA elements of phenobarbital-responsive cytochrome P450 CYP2B genes. J Biochem Mol Toxicol 12: 3–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
