

Conserved modular domains team up to latch-open active protein kinase Cα
- PMID: 24790081
- PMCID: PMC4067214
- DOI: 10.1074/jbc.M113.534750
Conserved modular domains team up to latch-open active protein kinase Cα
Abstract
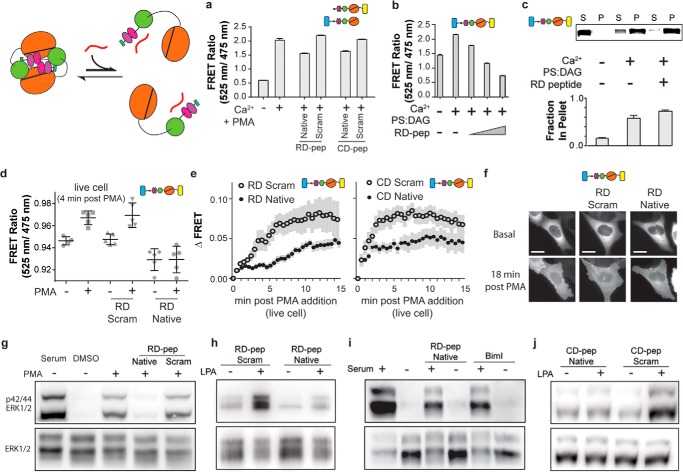
Signaling proteins comprised of modular domains have evolved along with multicellularity as a method to facilitate increasing intracellular bandwidth. The effects of intramolecular interactions between modular domains within the context of native proteins have been largely unexplored. Here we examine intra- and intermolecular interactions in the multidomain signaling protein, protein kinase Cα (PKCα). We identify three interactions between two activated PKC molecules that synergistically stabilize a nanomolar affinity homodimer. Disruption of the homodimer results in a loss of PKC-mediated ERK1/2 phosphorylation, whereas disruption of the auto-inhibited state promotes the homodimer and prolongs PKC membrane localization. These observations support a novel regulatory mechanism wherein homodimerization dictates the equilibrium between the auto-inhibited and active states of PKC by sequestering auto-inhibitory interactions. Our findings underscore the physiological importance of context-dependent modular domain interactions in cell signaling.
Keywords: Extracellular Signal-regulated Kinase (ERK); Fluorescence Resonance Energy Transfer (FRET); Homodimerization; Modular Domains; PKC; Protein Domain; Protein Kinase C (PKC); Protein-Protein Interaction.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures










References
-
- Copley R. R., Schultz J., Ponting C. P., Bork P. (1999) Protein families in multicellular organisms. Curr. Opin. Struct. Biol. 9, 408–415 - PubMed
-
- Lander E. S., et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921 - PubMed
-
- Pawson T., Nash P. (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300, 445–452 - PubMed
-
- Bhattacharyya R. P., Reményi A., Yeh B. J., Lim W. A. (2006) Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 75, 655–680 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
