

Distinct functional roles for the two SLX4 ubiquitin-binding UBZ domains mutated in Fanconi anemia
- PMID: 24794496
- PMCID: PMC4075355
- DOI: 10.1242/jcs.146167
Distinct functional roles for the two SLX4 ubiquitin-binding UBZ domains mutated in Fanconi anemia
Abstract
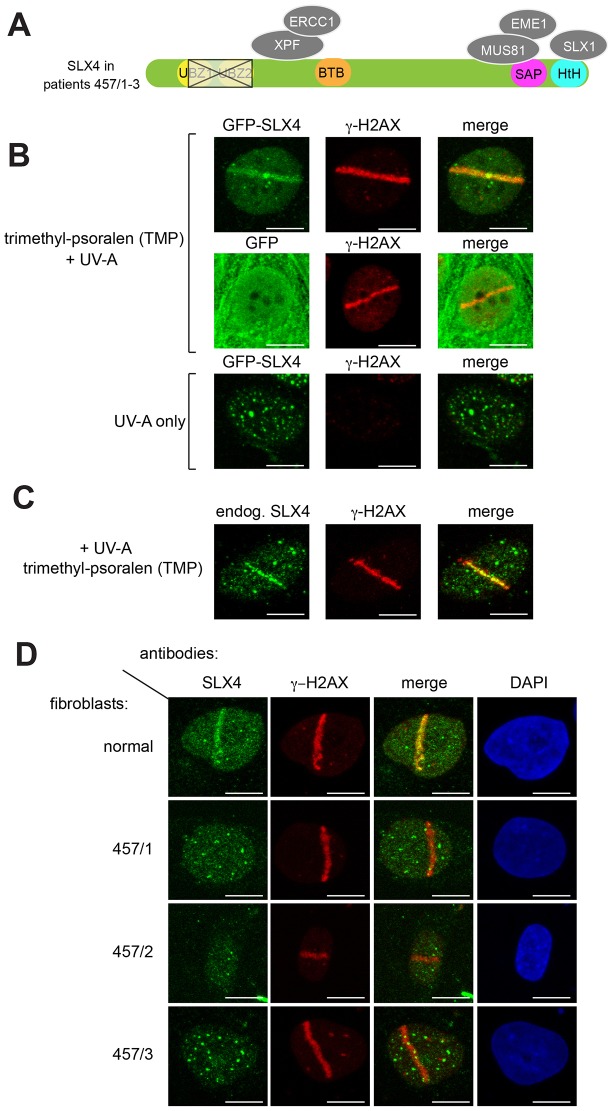
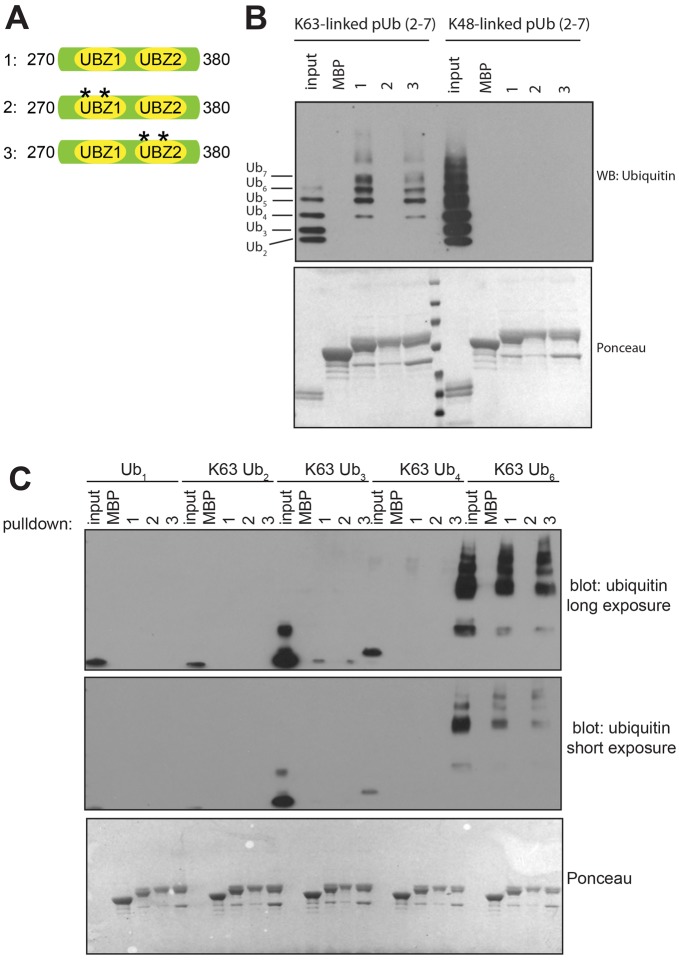
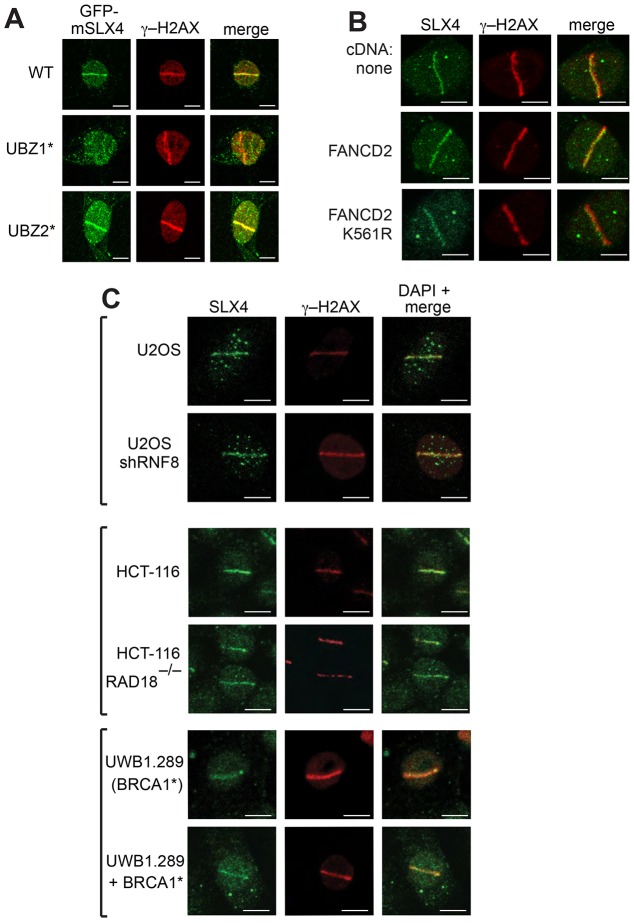
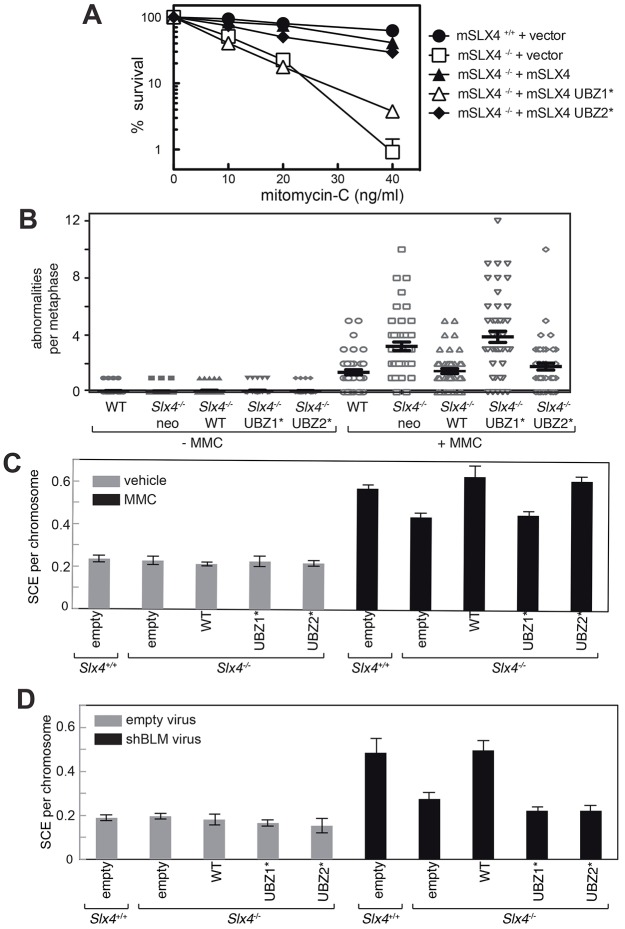
Defects in SLX4, a scaffold for DNA repair nucleases, result in Fanconi anemia (FA), due to the defective repair of inter-strand DNA crosslinks (ICLs). Some FA patients have an SLX4 deletion removing two tandem UBZ4-type ubiquitin-binding domains that are implicated in protein recruitment to sites of DNA damage. Here, we show that human SLX4 is recruited to sites of ICL induction but that the UBZ-deleted form of SLX4 in cells from FA patients is not. SLX4 recruitment does not require either the ubiquitylation of FANCD2 or the E3 ligases RNF8, RAD18 and BRCA1. We show that the first (UBZ-1) but not the second UBZ domain of SLX4 binds to ubiquitin polymers, with a preference for K63-linked chains. Furthermore, UBZ-1 is required for SLX4 recruitment to ICL sites and for efficient ICL repair in murine fibroblasts. The SLX4 UBZ-2 domain does not bind to ubiquitin in vitro or contribute to ICL repair, but it is required for the resolution of Holliday junctions in vivo. These data shed light on SLX4 recruitment, and they point to the existence of currently unidentified ubiquitylated ligands and E3 ligases that are crucial for ICL repair.
Keywords: FANCP; Fanconi anemia; ICL; SLX4; UBZ; Ubiquitin.
© 2014. Published by The Company of Biologists Ltd.
Figures
References
-
- Fekairi S., Scaglione S., Chahwan C., Taylor E. R., Tissier A., Coulon S., Dong M. Q., Ruse C., Yates J. R., III, Russell P. et al.(2009). Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89 10.1016/j.cell.2009.06.029 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
