

Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype
- PMID: 24794859
- PMCID: PMC4140467
- DOI: 10.1093/hmg/ddu201
Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype
Abstract
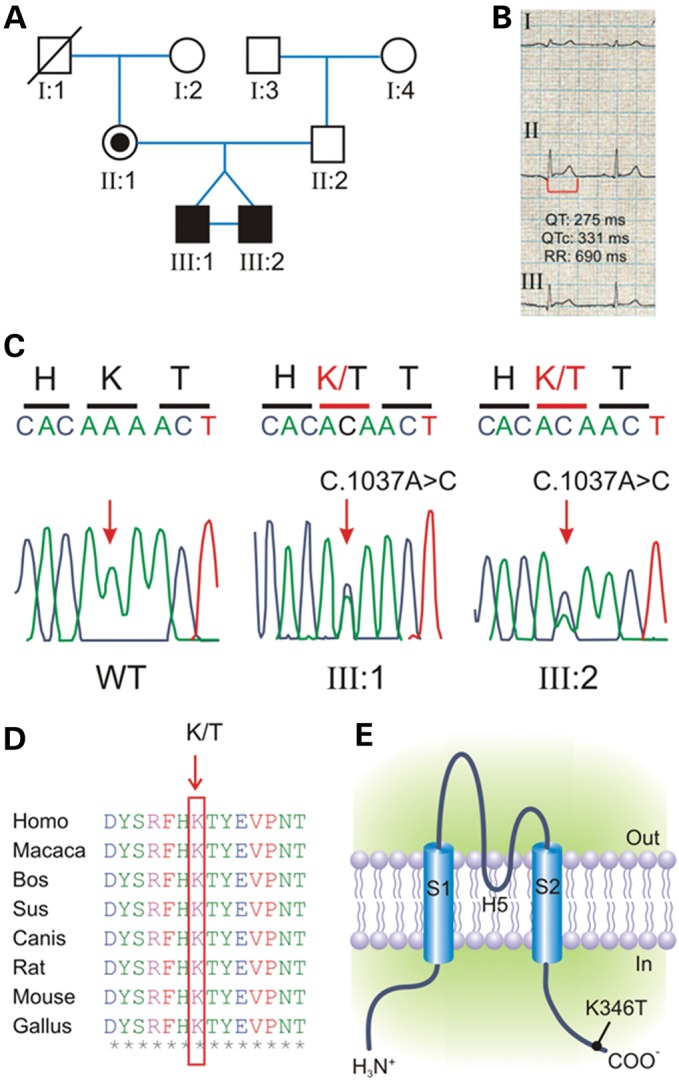
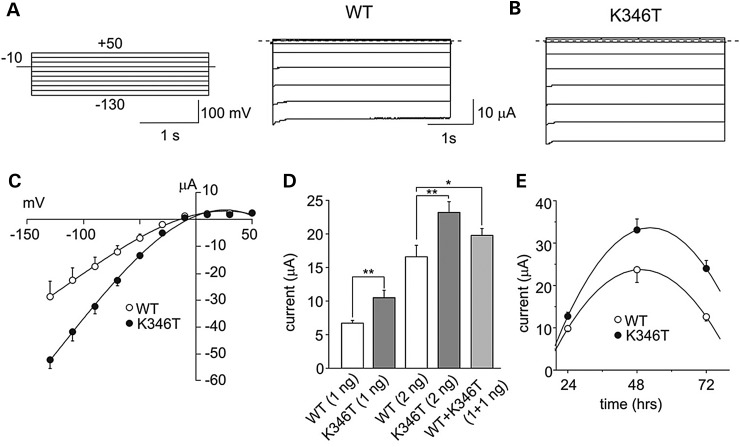
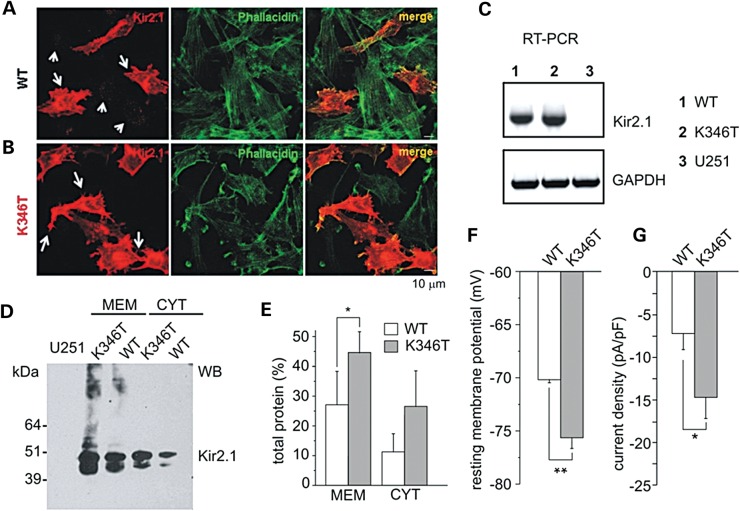
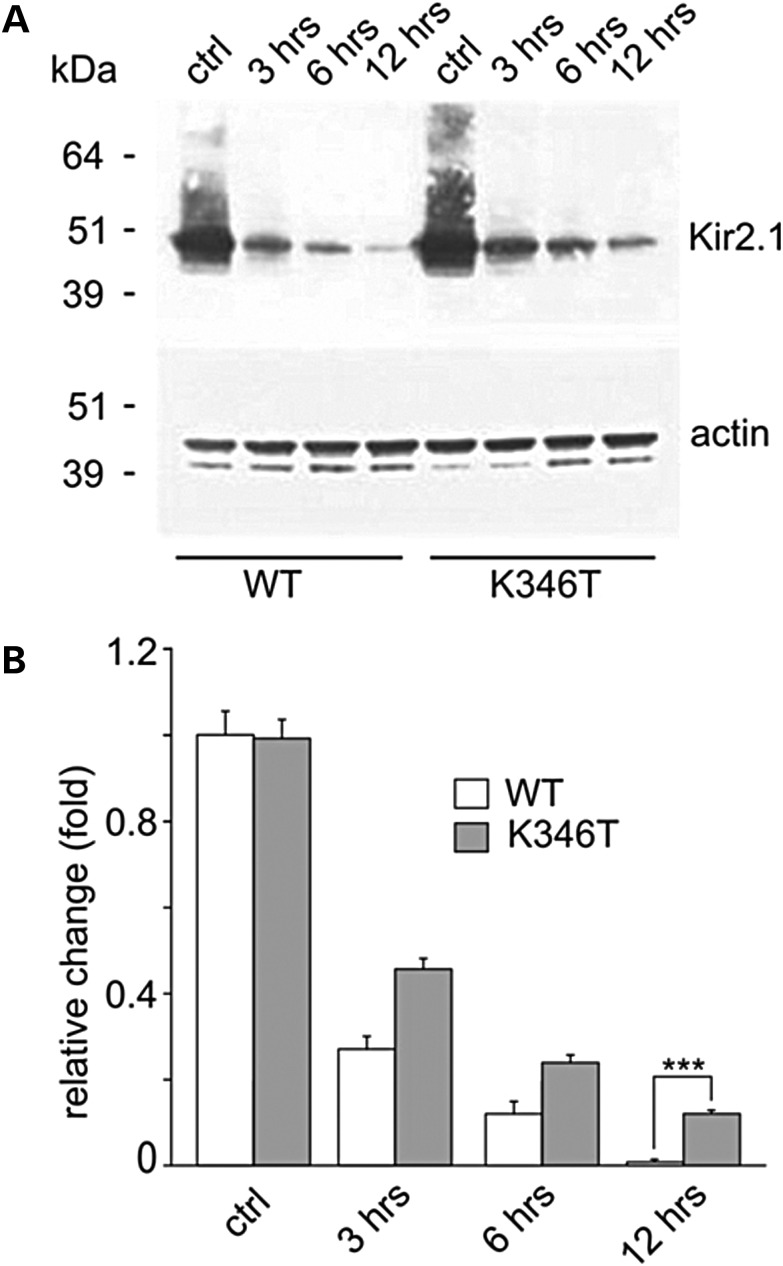
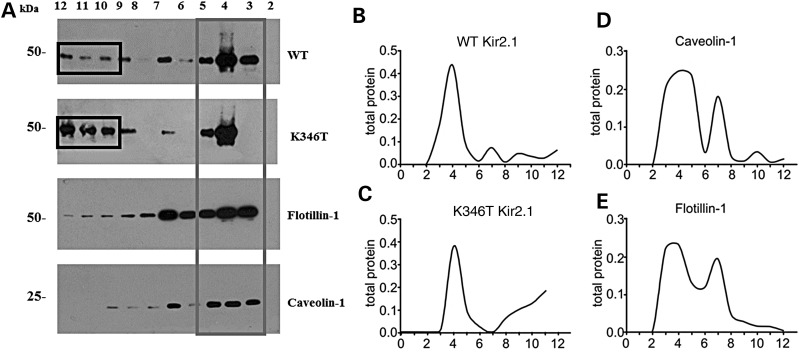
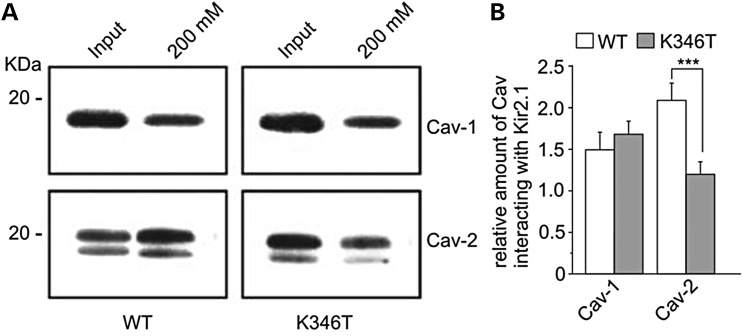
Short QT3 syndrome (SQT3S) is a cardiac disorder characterized by a high risk of mortality and associated with mutations in Kir2.1 (KCNJ2) channels. The molecular mechanisms leading to channel dysfunction, cardiac rhythm disturbances and neurodevelopmental disorders, potentially associated with SQT3S, remain incompletely understood. Here, we report on monozygotic twins displaying a short QT interval on electrocardiogram recordings and autism-epilepsy phenotype. Genetic screening identified a novel KCNJ2 variant in Kir2.1 that (i) enhanced the channel's surface expression and stability at the plasma membrane, (ii) reduced protein ubiquitylation and degradation, (iii) altered protein compartmentalization in lipid rafts by targeting more channels to cholesterol-poor domains and (iv) reduced interactions with caveolin 2. Importantly, our study reveals novel physiological mechanisms concerning wild-type Kir2.1 channel processing by the cell, such as binding to both caveolin 1 and 2, protein degradation through the ubiquitin-proteasome pathway; in addition, it uncovers a potential multifunctional site that controls Kir2.1 surface expression, protein half-life and partitioning to lipid rafts. The reported mechanisms emerge as crucial also for proper astrocyte function, suggesting the need for a neuropsychiatric evaluation in patients with SQT3S and offering new opportunities for disease management.
© The Author 2014. Published by Oxford University Press.
Figures
References
-
- Yoon G., Quitania L., Kramer J.H., Fu Y.H., Miller B.L., Ptácek L.J. Andersen-Tawil syndrome, definition of a neurocognitive phenotype. Neurology. 2006;66:1703–1710. - PubMed
-
- Cheung C.L., Lau K.S., Ho A.Y., Lee K.K., Tiu S.C., Lau E.Y., Leung J., Tsang M.W., Chan K.W., Yeung C.Y., et al. Genome-wide association study identifies a susceptibility locus for thyrotoxic periodic paralysis at 17q24.3. Nat. Genet. 2012;44:1026–1029. - PubMed
-
- Chan H.F., Chen M.L., Su J.J., Ko L.C., Lin C.H., Wu R.M. A novel neuropsychiatric phenotype of KCNJ2 mutation in one Taiwanese family with Andersen-Tawil syndrome. J. Hum. Genet. 2010;55:186–188. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
