

A mutation causing Brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents
- PMID: 24795344
- PMCID: PMC4114079
- DOI: 10.1161/CIRCGENETICS.113.000480
A mutation causing Brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents
Abstract
Background: The mechanisms of the electrocardiographic changes and arrhythmias in Brugada syndrome (BrS) remain controversial. Mutations in the sodium channel gene, SCN5A, and regulatory proteins that reduce or eliminate sodium current (INa) have been linked to BrS. We studied the properties of a BrS-associated SCN5A mutation in a protein kinase A (PKA) consensus phosphorylation site, R526H.
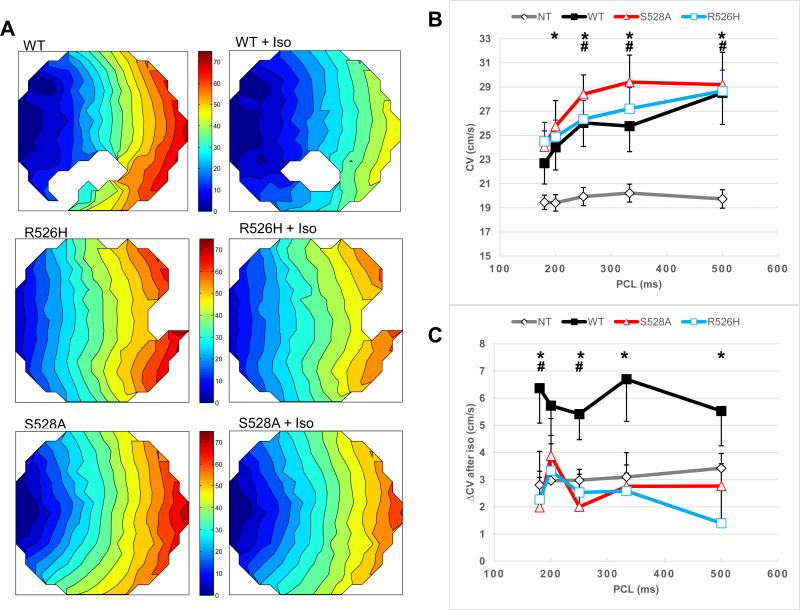
Methods and results: In vitro PKA phosphorylation was detected in the I-II linker peptide of wild-type (WT) channels but not R526H or S528A (phosphorylation site) mutants. Cell surface expression of R526H and S528A channels was reduced compared with WT. Whole-cell INa through all channel variants revealed no significant differences in the steady-state activation, inactivation, and recovery from inactivation. Peak current densities of the mutants were significantly reduced compared with WT. Infection of 2D cultures of neonatal rat ventricular myocytes with WT and mutant channels increased conduction velocity compared with noninfected cells. PKA stimulation significantly increased peak INa and conduction velocity of WT but not mutant channels. Oxidant stress inhibits cardiac INa; WT and mutant INa decreases with the intracellular application of reduced nicotinamide adenine dinucleotide (NADH), an effect that is reversed by PKA stimulation in WT but not in R526H or S528A channels.
Conclusions: We identified a family with BrS and an SCN5A mutation in a PKA consensus phosphorylation site. The BrS mutation R526H is associated with a reduction in the basal level of INa and a failure of PKA stimulation to augment the current that may contribute to the predisposition to arrhythmias in patients with BrS, independent of the precipitants.
Keywords: death, sudden, cardiac; ion channel; mutation; reactive oxygen species.
© 2014 American Heart Association, Inc.
Figures

References
-
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: Report of the second consensus conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. - PubMed
-
- Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, et al. Ventricular fibrillation without apparent heart disease: Description of six cases. Am Heart J. 1989;118:1203–1209. - PubMed
-
- Haissaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–2023. - PubMed
-
- Hoogendijk MG, Opthof T, Postema PG, Wilde AA, de Bakker JM, Coronel R. The Brugada ECG pattern: A marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol. 2010;3:283–290. - PubMed
-
- Patel SS, Anees S, Ferrick KJ. Prevalence of a Brugada pattern electrocardiogram in an urban population in the United States. Pacing Clin Electrophysiol. 2009;32:704–708. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
