

Structure-energy-based predictions and network modelling of RASopathy and cancer missense mutations
- PMID: 24803665
- PMCID: PMC4188041
- DOI: 10.1002/msb.20145092
Structure-energy-based predictions and network modelling of RASopathy and cancer missense mutations
Abstract
The Ras/MAPK syndromes ('RASopathies') are a class of developmental disorders caused by germline mutations in 15 genes encoding proteins of the Ras/mitogen-activated protein kinase (MAPK) pathway frequently involved in cancer. Little is known about the molecular mechanisms underlying the differences in mutations of the same protein causing either cancer or RASopathies. Here, we shed light on 956 RASopathy and cancer missense mutations by combining protein network data with mutational analyses based on 3D structures. Using the protein design algorithm FoldX, we predict that most of the missense mutations with destabilising energies are in structural regions that control the activation of proteins, and only a few are predicted to compromise protein folding. We find a trend that energy changes are higher for cancer compared to RASopathy mutations. Through network modelling, we show that partly compensatory mutations in RASopathies result in only minor downstream pathway deregulation. In summary, we suggest that quantitative rather than qualitative network differences determine the phenotypic outcome of RASopathy compared to cancer mutations.
Figures
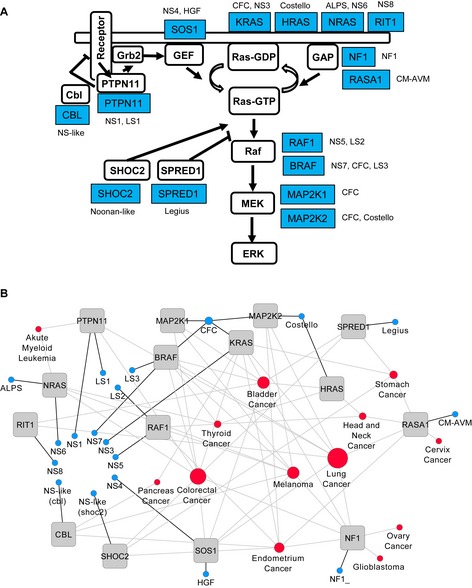
Network diagram showing affected genes in
RAS opathies. Proteins (PTPN11, SOS1 andSPRED ) and protein groups (Ras, includingNRAS ,HRAS ,KRAS and RIT1;GAP , including NF1 and RASA1; Raf, including RAF1 and BRAF; MEK, includingMAP 2K1 and MAP2K2) are displayed in white boxes and arranged in a network with their respective genes in grey. RASopathy diseases are indicated in blue.Diseasome of RASopathies and cancer. Each node corresponds to a distinct disorder or cancer type. The size of the node corresponds to the total number of genes (among the 15 genes) that are involved in a particular disease. Abbreviations: NS, Noonan syndrome; NF1, neurofibromatosis type 1;
CFC , cardiofaciocutaneous;LS ,LEOPARD syndrome; HGF, hereditary gingvial fibromatosis; CM‐AVM, capillary malfunction‐arteriovenous malfunction;ALPS , autoimmune lymphoproliferative syndrome. Suffixes in NS (NS1, NS3, NS4, NS5, NS6, NS7, NS8 and NS‐like) and LS (LS1 to LS3) are different forms of the respective disease according to the classification in theOMIM database.
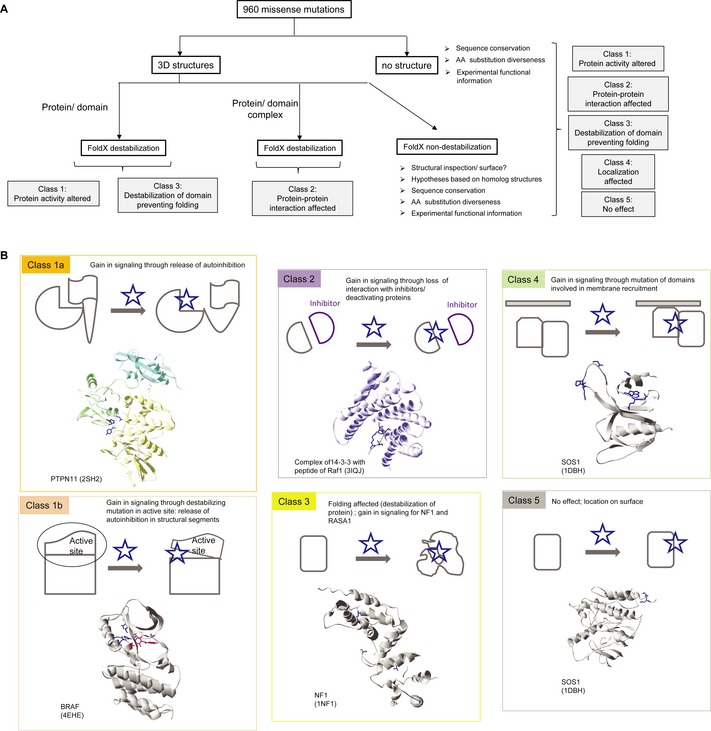
Flow chart of the analysis performed in this work.
Classification of missense mutations. Class 1a mutants can impair domain‐domain inhibitory interactions, as found in the phosphatase
PTPN 11 or the GEF Sos1. Class 1b mutants may release an inhibitory protein segment, thus resulting in activation, for example mutations in activation segment in kinases, such asBRAF . Class 2 mutants affect protein‐protein interactions and can result in a gain in signalling results from the loss of interactions with inhibitors or deactivating proteins. Examples are RAS mutations that prevent the down‐regulation by RASA1, or the binding of 14‐3‐3 proteins, which has been shown to interfere with Ras binding and inhibit Ras‐mediated plasma membrane recruitment of RAF1. Class 3 mutants destabilise a globular domain preventing folding. Class 4 mutants can affect protein localisation and/or half‐life. Class 5 mutants are neutral and are often localised on the protein surface.
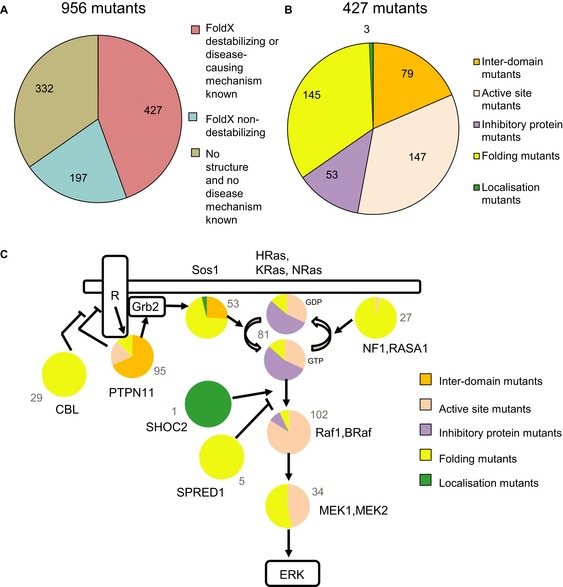
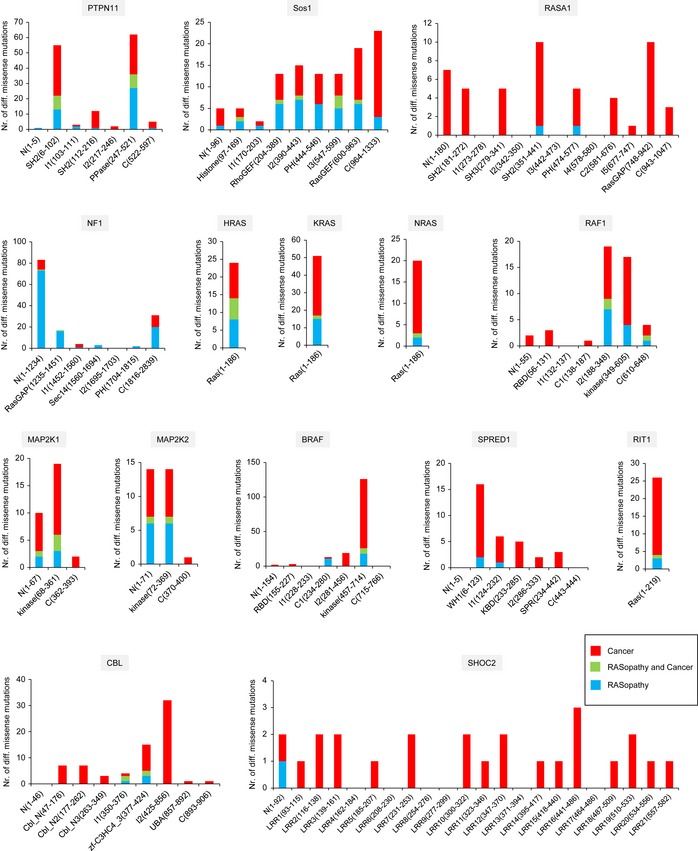
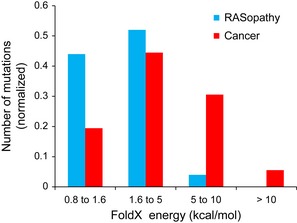
Structural coverage and the number of mutations predicted to be destabilising and non‐destabilising by FoldX.
Distribution of 427 missense mutations that map to a 3D structure into different classes.
Summary of the predicted effect for the 14 disease genes (proteins represented as pie diagrams). Proteins are arranged in a network similar as in Fig 1A. The grey numbers indicate the total number of different mutations represented in the pie diagrams.
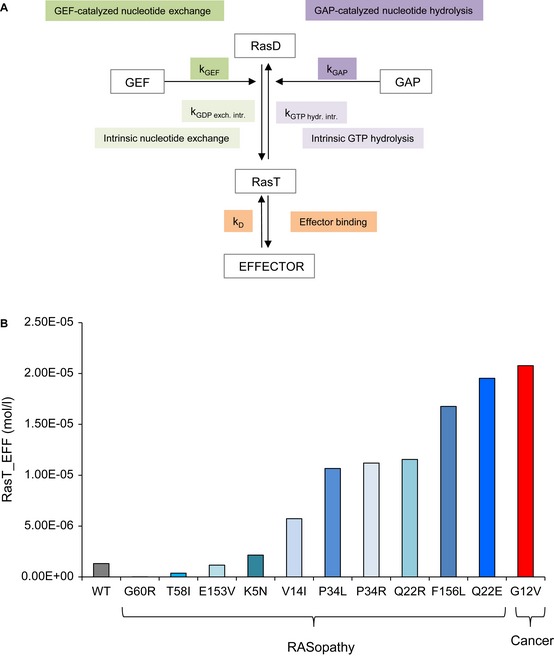
Schematic representation of the network model. The 5 indicated rate constants represent the five experimentally measured rate constants for Ras WT and mutants. kGEF is the
GEF catalysedGDP dissociation, kGAP is the GAP‐catalysed GTP hydrolysis, kGDP exch. intr. is the intrinsic nucleotide dissociation, kGTP hydr. intr. is the intrinsic GTP hydrolysis, and kD is the affinity between Ras binding to the effector (EFF).Results of the amount of Ras binding to effectors (Ras_EFF) in equilibrium for 12 network model simulations for using the respective 5 rate constants for Ras WT, Ras G12V (cancer) and RASopathy mutations (Ras K5N, V14I, Q22E, Q22R, P34L, P34R, T58I, G60R, E153V, F156L).
References
-
- Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, Kamata T, Lee P, Williams A, McMahon M, Marais R, Pritchard C (2012) The intermediate‐activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev 26: 1945–1958 - PMC - PubMed
-
- Aoki Y, Niihori T, Narumi Y, Kure S, Matsubara Y (2008) The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat 29: 992–1006 - PubMed
-
- Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, Ogata T, Takada F, Yano M, Ando T, Hoshika T, Barnett C, Ohashi H, Kawame H, Hasegawa T, Okutani T, Nagashima T, Hasegawa S, Funayama R, Nakayama K et al (2013) Gain‐of‐function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet 93: 173–180 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
