

Evolution of mitochondria reconstructed from the energy metabolism of living bacteria
- PMID: 24804722
- PMCID: PMC4013037
- DOI: 10.1371/journal.pone.0096566
Evolution of mitochondria reconstructed from the energy metabolism of living bacteria
Abstract
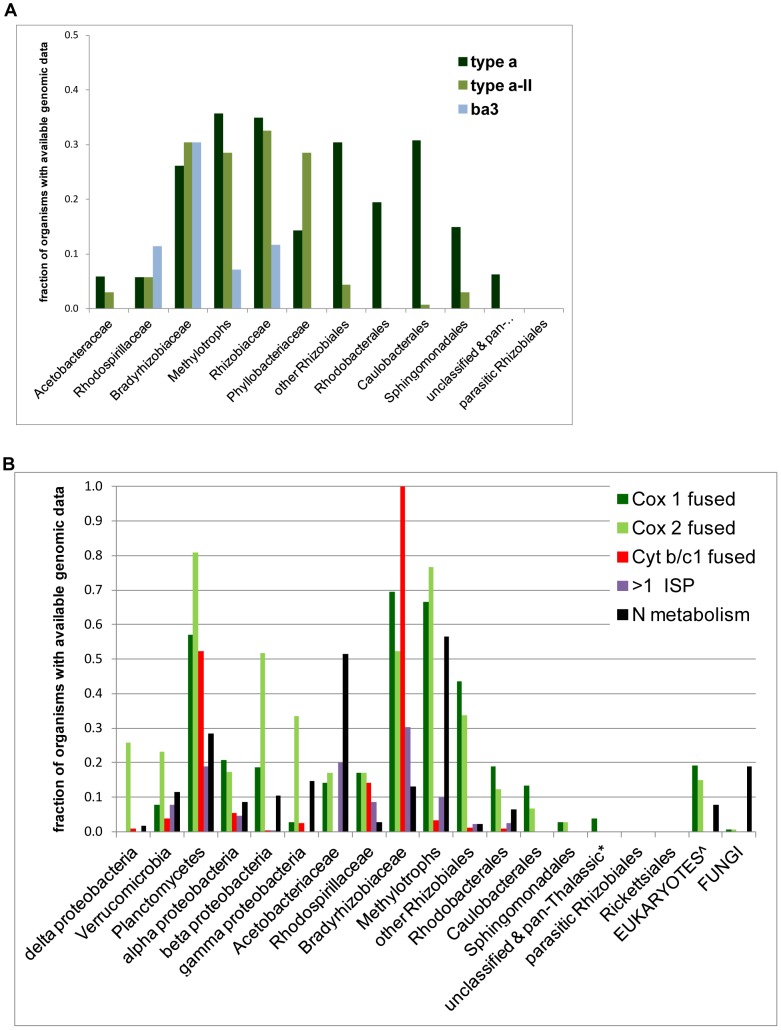
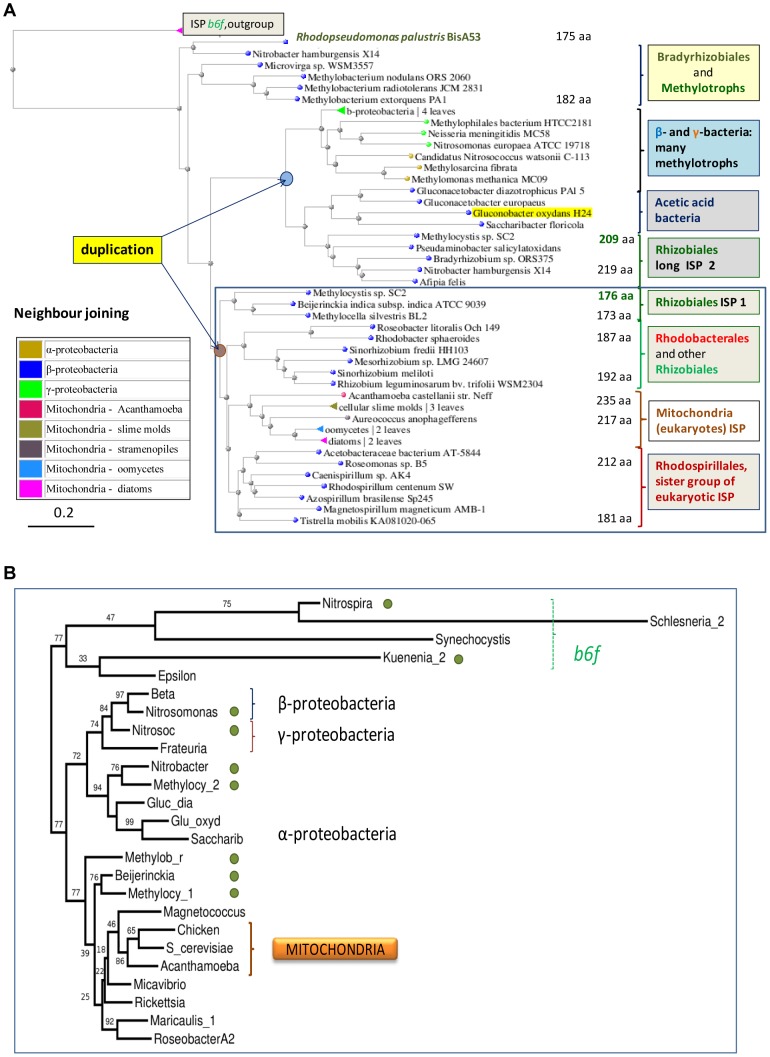
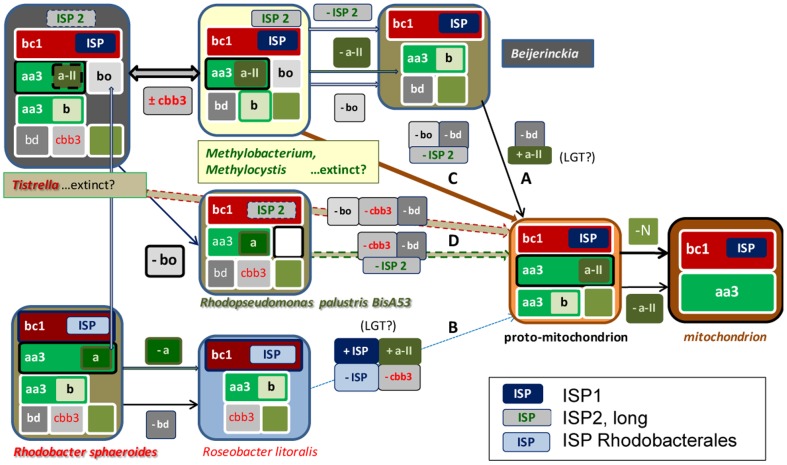
The ancestors of mitochondria, or proto-mitochondria, played a crucial role in the evolution of eukaryotic cells and derived from symbiotic α-proteobacteria which merged with other microorganisms - the basis of the widely accepted endosymbiotic theory. However, the identity and relatives of proto-mitochondria remain elusive. Here we show that methylotrophic α-proteobacteria could be the closest living models for mitochondrial ancestors. We reached this conclusion after reconstructing the possible evolutionary pathways of the bioenergy systems of proto-mitochondria with a genomic survey of extant α-proteobacteria. Results obtained with complementary molecular and genetic analyses of diverse bioenergetic proteins converge in indicating the pathway stemming from methylotrophic bacteria as the most probable route of mitochondrial evolution. Contrary to other α-proteobacteria, methylotrophs show transition forms for the bioenergetic systems analysed. Our approach of focusing on these bioenergetic systems overcomes the phylogenetic impasse that has previously complicated the search for mitochondrial ancestors. Moreover, our results provide a new perspective for experimentally re-evolving mitochondria from extant bacteria and in the future produce synthetic mitochondria.
Conflict of interest statement
Figures
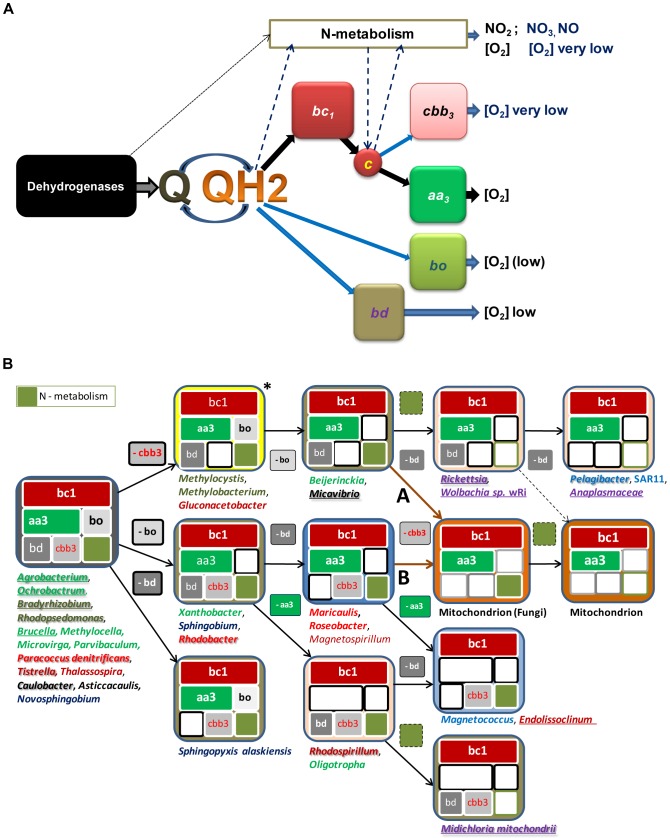
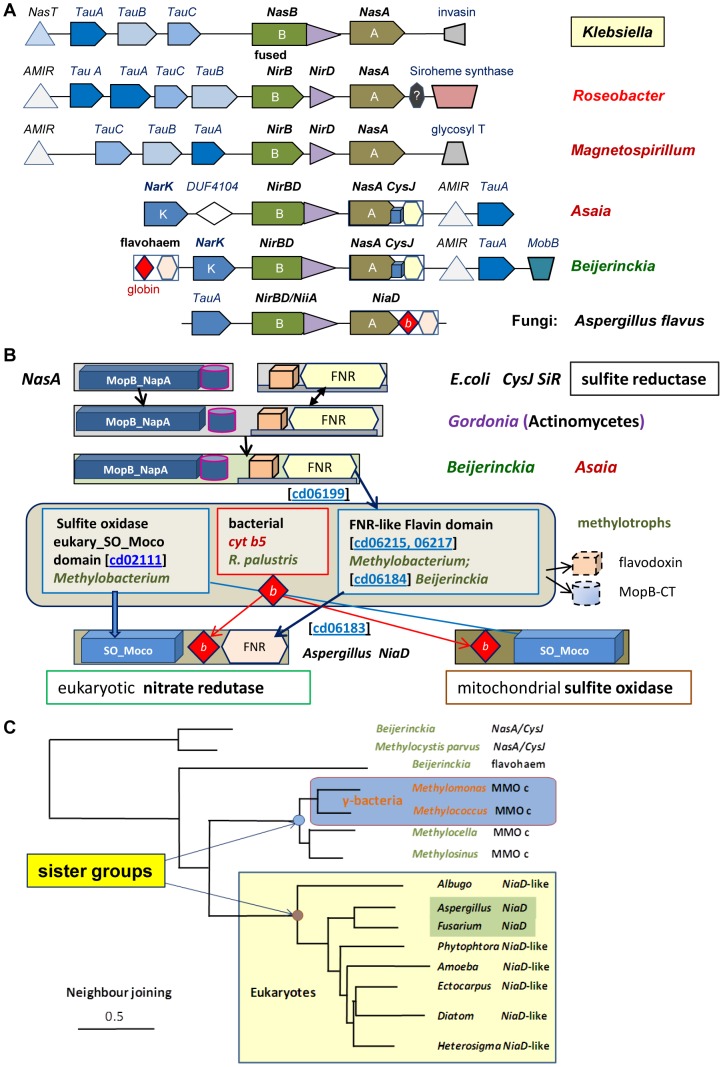
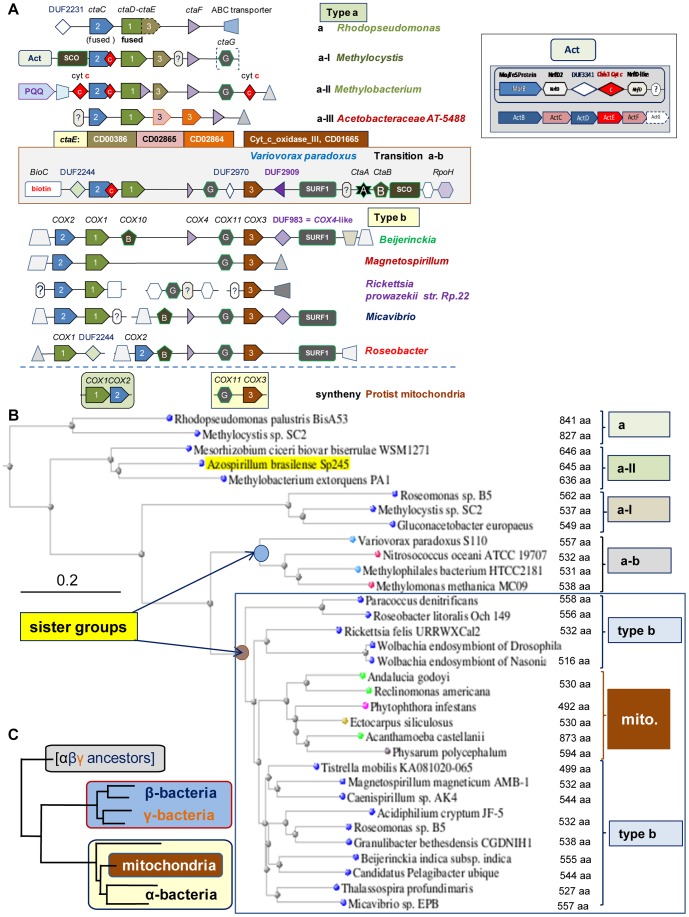

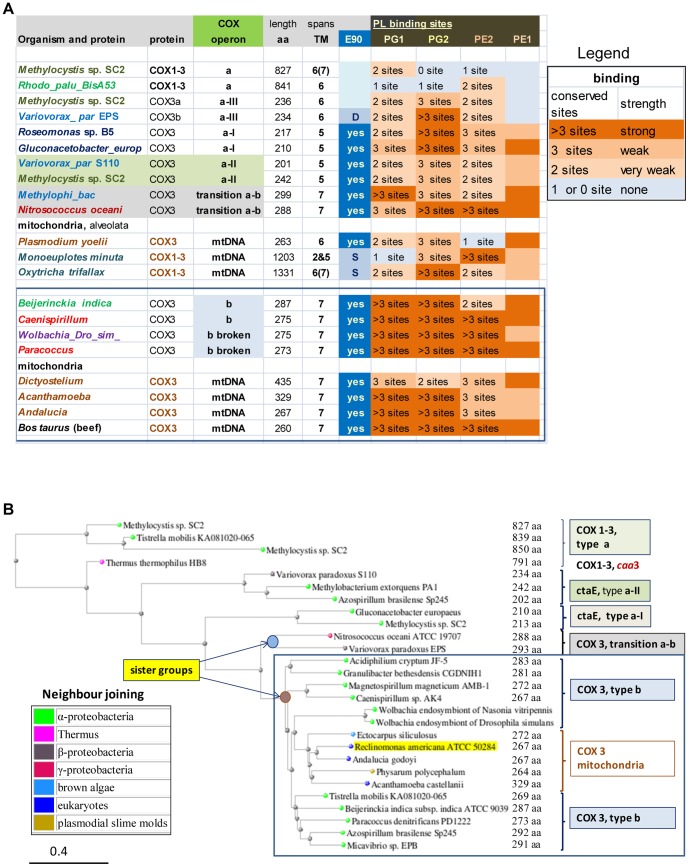

Similar articles
-
Bioenergetic evolution in proteobacteria and mitochondria.Genome Biol Evol. 2014 Nov 27;6(12):3238-51. doi: 10.1093/gbe/evu257. Genome Biol Evol. 2014. PMID: 25432941 Free PMC article.
-
Rickettsiaceae, rickettsia-like endosymbionts, and the origin of mitochondria.Biosci Rep. 2001 Feb;21(1):1-17. doi: 10.1023/a:1010409415723. Biosci Rep. 2001. PMID: 11508688 Review.
-
Phylogenomic reconstruction indicates mitochondrial ancestor was an energy parasite.PLoS One. 2014 Oct 15;9(10):e110685. doi: 10.1371/journal.pone.0110685. eCollection 2014. PLoS One. 2014. PMID: 25333787 Free PMC article.
-
Single eubacterial origin of eukaryotic sulfide:quinone oxidoreductase, a mitochondrial enzyme conserved from the early evolution of eukaryotes during anoxic and sulfidic times.Mol Biol Evol. 2003 Sep;20(9):1564-74. doi: 10.1093/molbev/msg174. Epub 2003 Jun 27. Mol Biol Evol. 2003. PMID: 12832624
-
On the origin of mitochondria: a genomics perspective.Philos Trans R Soc Lond B Biol Sci. 2003 Jan 29;358(1429):165-77; discussion 177-9. doi: 10.1098/rstb.2002.1193. Philos Trans R Soc Lond B Biol Sci. 2003. PMID: 12594925 Free PMC article. Review.
Cited by
-
Alpha proteobacterial ancestry of the [Fe-Fe]-hydrogenases in anaerobic eukaryotes.Biol Direct. 2016 Jul 30;11:34. doi: 10.1186/s13062-016-0136-3. Biol Direct. 2016. PMID: 27473689 Free PMC article.
-
Cell death and DNA damage via ROS mechanisms after applied antibiotics and antioxidants doses in prostate hyperplasia primary cell cultures.Medicine (Baltimore). 2024 Sep 13;103(37):e39450. doi: 10.1097/MD.0000000000039450. Medicine (Baltimore). 2024. PMID: 39287312 Free PMC article.
-
Lactobacillus rhamnosus GG Modulates Mitochondrial Function and Antioxidant Responses in an Ethanol-Exposed In Vivo Model: Evidence of HIGD2A-Dependent OXPHOS Remodeling in the Liver.Antioxidants (Basel). 2025 May 23;14(6):627. doi: 10.3390/antiox14060627. Antioxidants (Basel). 2025. PMID: 40563263 Free PMC article.
-
Energy metabolism in anaerobic eukaryotes and Earth's late oxygenation.Free Radic Biol Med. 2019 Aug 20;140:279-294. doi: 10.1016/j.freeradbiomed.2019.03.030. Epub 2019 Mar 29. Free Radic Biol Med. 2019. PMID: 30935869 Free PMC article. Review.
-
Evolution of the Inhibitory and Non-Inhibitory ε, ζ, and IF1 Subunits of the F1FO-ATPase as Related to the Endosymbiotic Origin of Mitochondria.Microorganisms. 2022 Jul 7;10(7):1372. doi: 10.3390/microorganisms10071372. Microorganisms. 2022. PMID: 35889091 Free PMC article. Review.
References
-
- Lane N, Martin W (2010) The energetics of genome complexity. Nature 467: 929–934. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
