

CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury
- PMID: 24806994
- PMCID: PMC4545982
- DOI: 10.1089/neu.2013.3252
CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury
Abstract
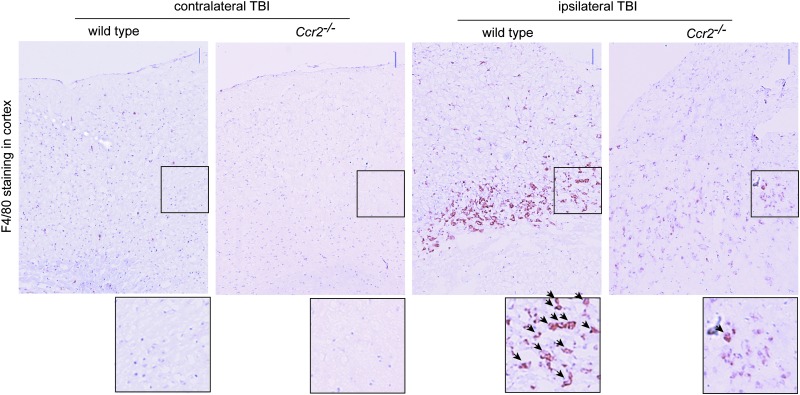
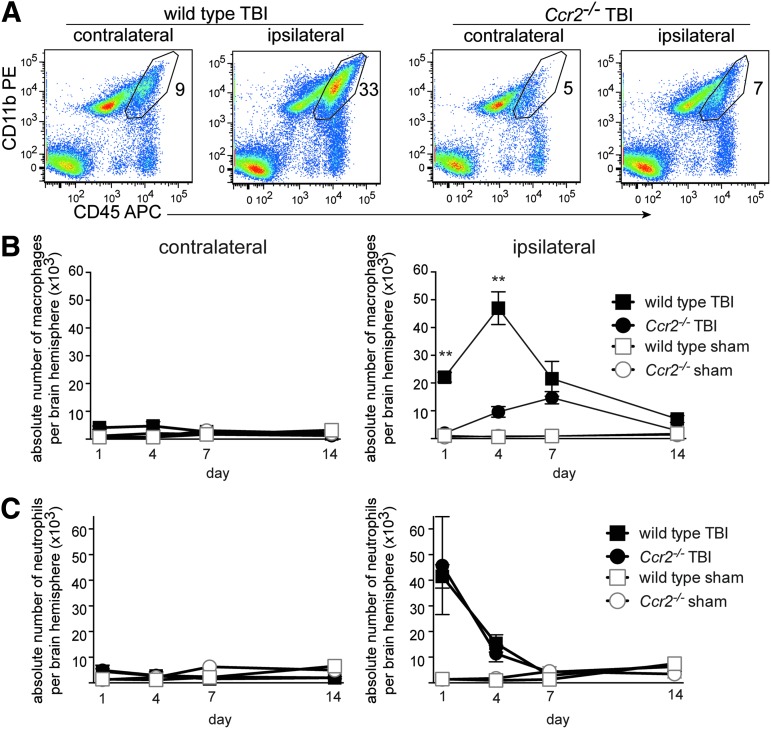
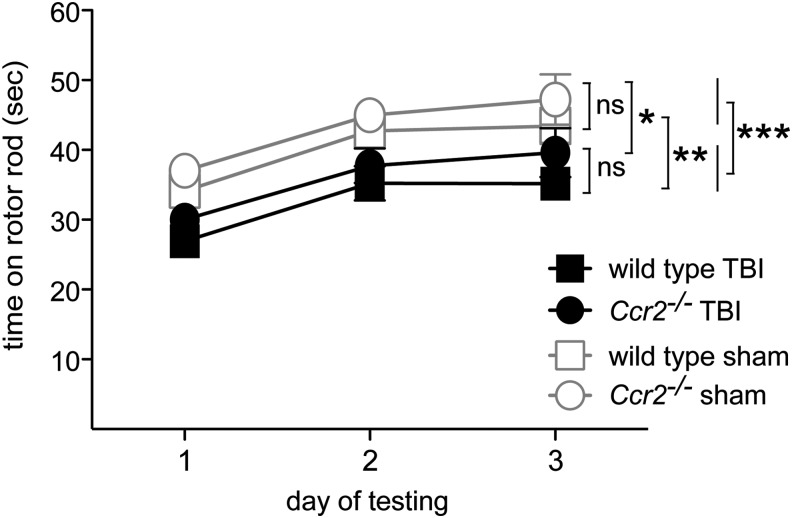
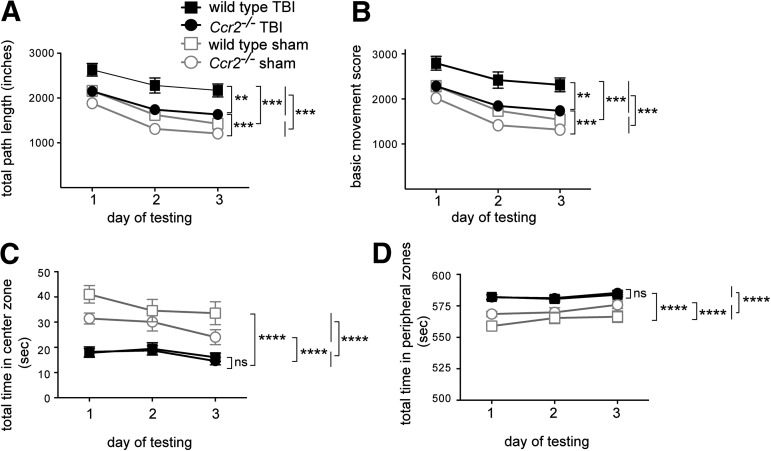
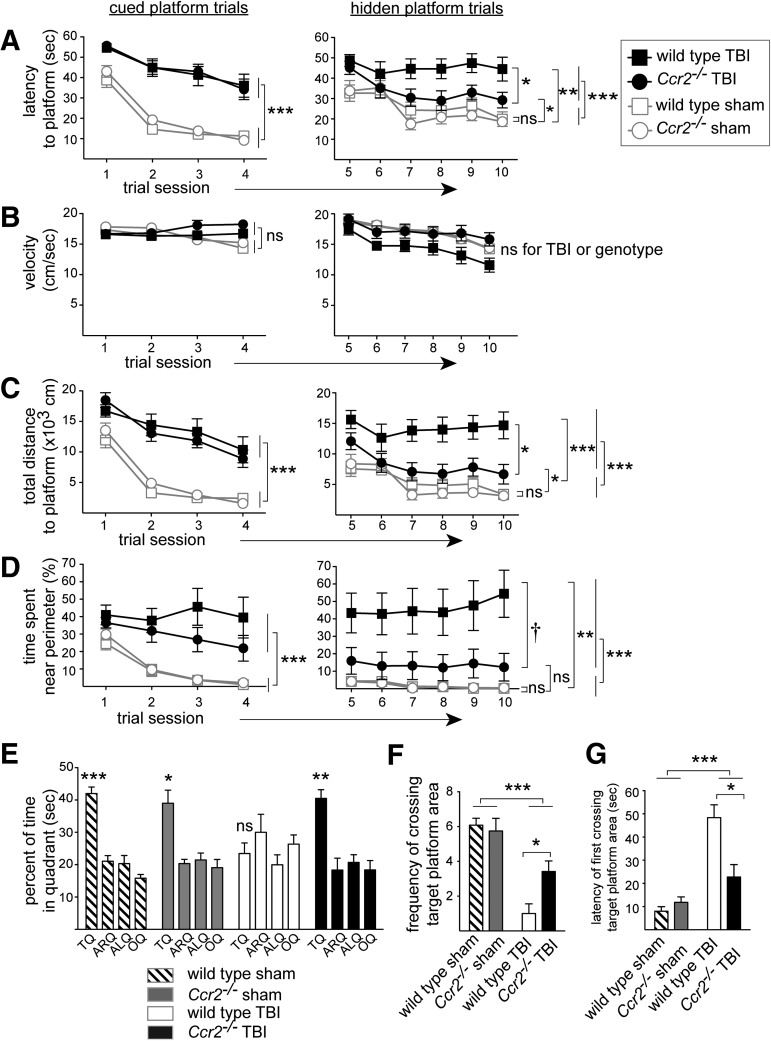
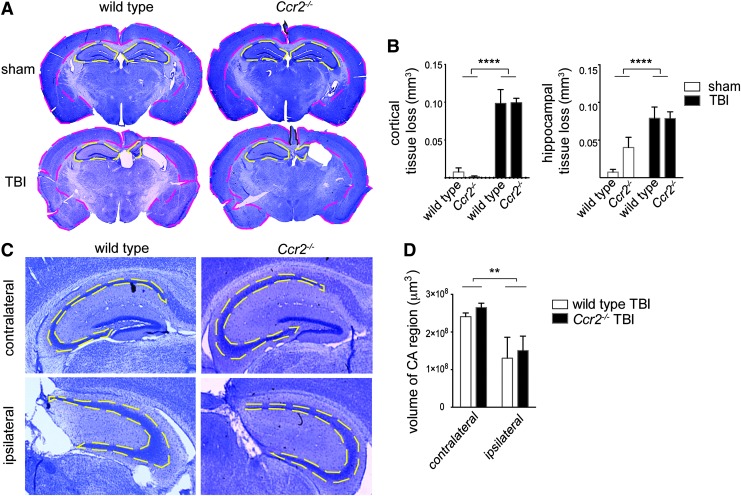
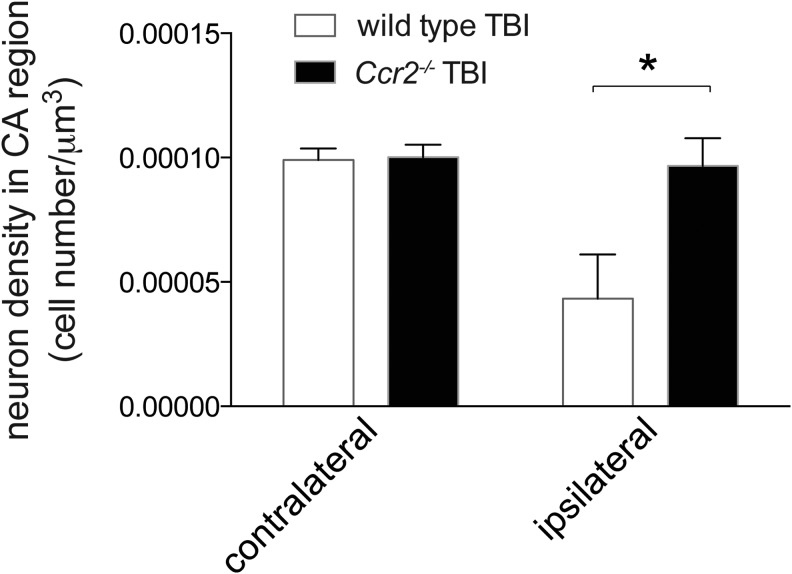
Traumatic brain injury (TBI) provokes inflammatory responses, including a dramatic rise in brain macrophages in the area of injury. The pathway(s) responsible for macrophage infiltration of the traumatically injured brain and the effects of macrophages on functional outcomes are not well understood. C-C-chemokine receptor 2 (CCR2) is known for directing monocytes to inflamed tissues. To assess the role of macrophages and CCR2 in TBI, we determined outcomes in CCR2-deficient (Ccr2(-/-)) mice in a controlled cortical impact model. We quantified brain myeloid cell numbers post-TBI by flow cytometry and found that Ccr2(-/-) mice had greatly reduced macrophage numbers (∼80-90% reduction) early post-TBI, compared with wild-type mice. Motor, locomotor, and cognitive outcomes were assessed. Lack of Ccr2 improved locomotor activity with less hyperactivity in open field testing, but did not affect anxiety levels or motor coordination on the rotarod three weeks after TBI. Importantly, Ccr2(-/-) mice demonstrated greater spatial learning and memory, compared with wild-type mice eight weeks after TBI. Although there was no difference in the volume of tissue loss, Ccr2(-/-) mice had significantly increased neuronal density in the CA1-CA3 regions of the hippocampus after TBI, compared with wild-type mice. These data demonstrate that Ccr2 directs the majority of macrophage homing to the brain early after TBI and indicates that Ccr2 may facilitate harmful responses. Lack of Ccr2 improves functional recovery and neuronal survival. These results suggest that therapeutic blockade of CCR2-dependent responses may improve outcomes following TBI.
Keywords: CCR2; brain injury; chemotaxis; inflammation; macrophage.
Figures
References
-
- Jain K.K. (2008). Neuroprotection in traumatic brain injury. Drug Discov. Today 13, 1082–1089 - PubMed
-
- Okie S. (2005). Traumatic brain injury in the war zone. N. Engl. J. Med. 352, 2043–2047 - PubMed
-
- Gomez C.R., Boehmer E.D., and Kovacs E.J. (2005). The aging innate immune system. Curr. Opin. Immunol. 17, 457–462 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
