

The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors
- PMID: 24806995
- PMCID: PMC4282530
- DOI: 10.1002/stem.1748
The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors
Abstract
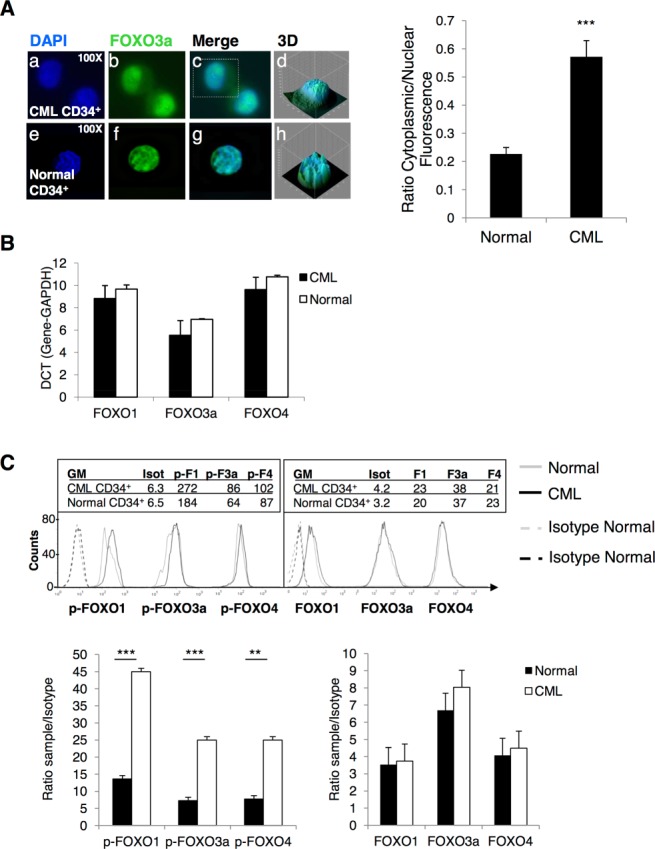
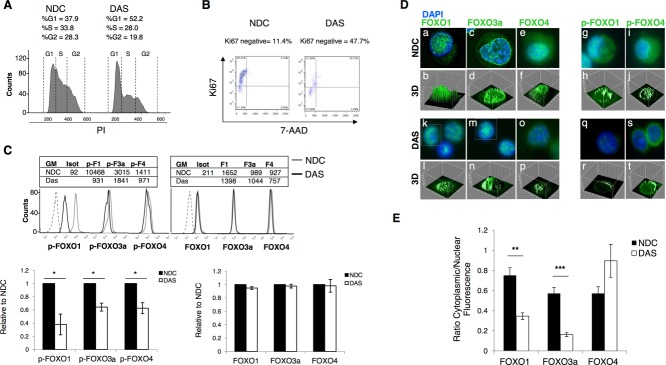
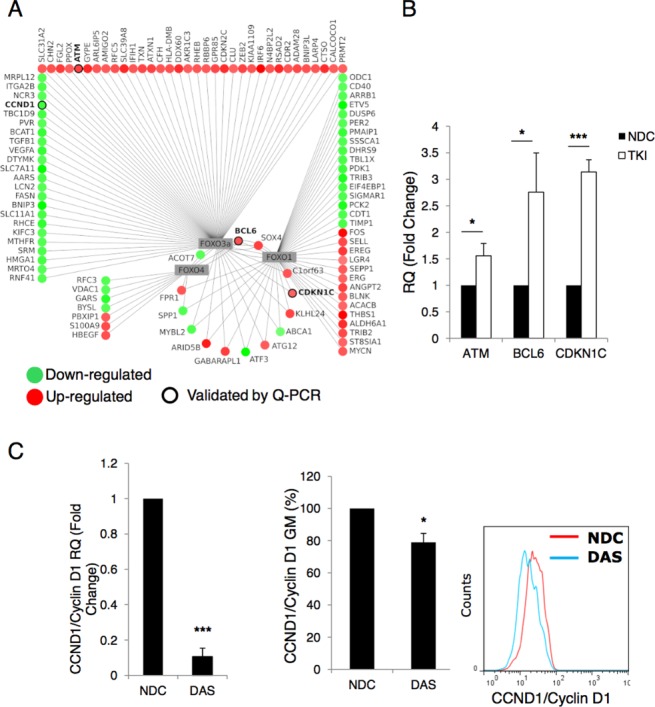
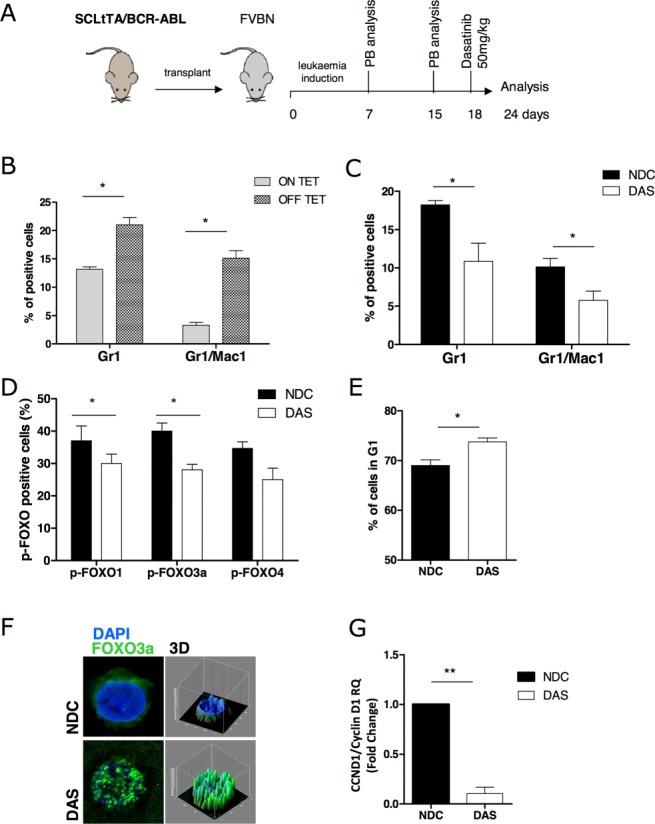
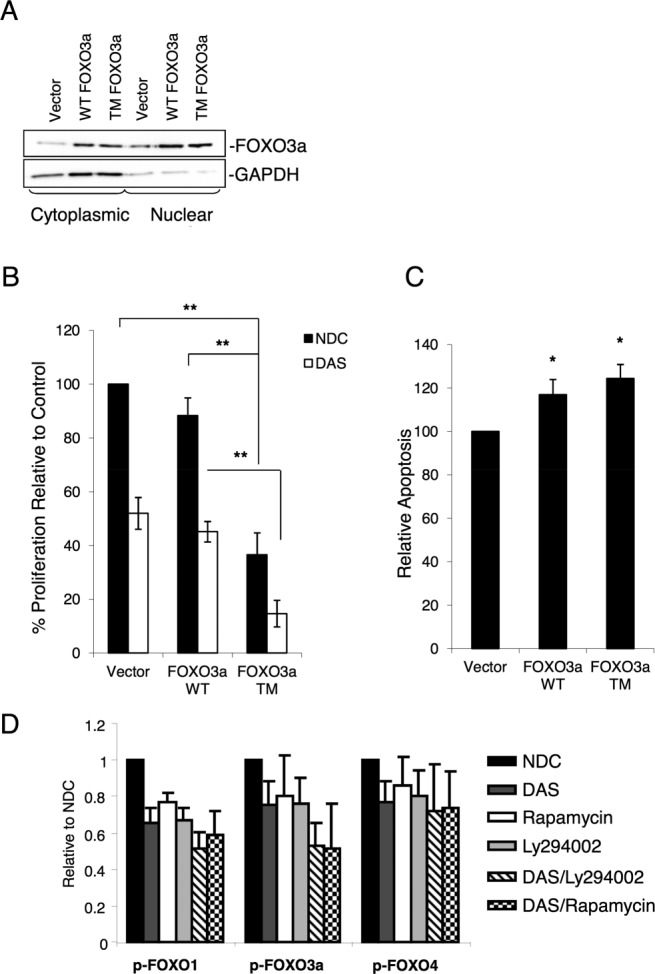
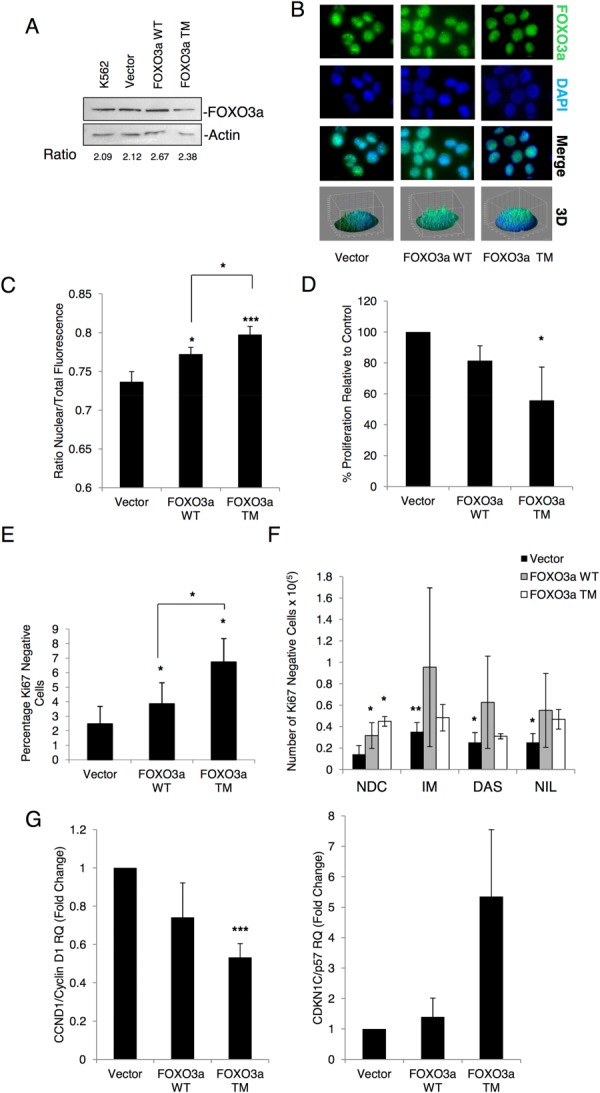
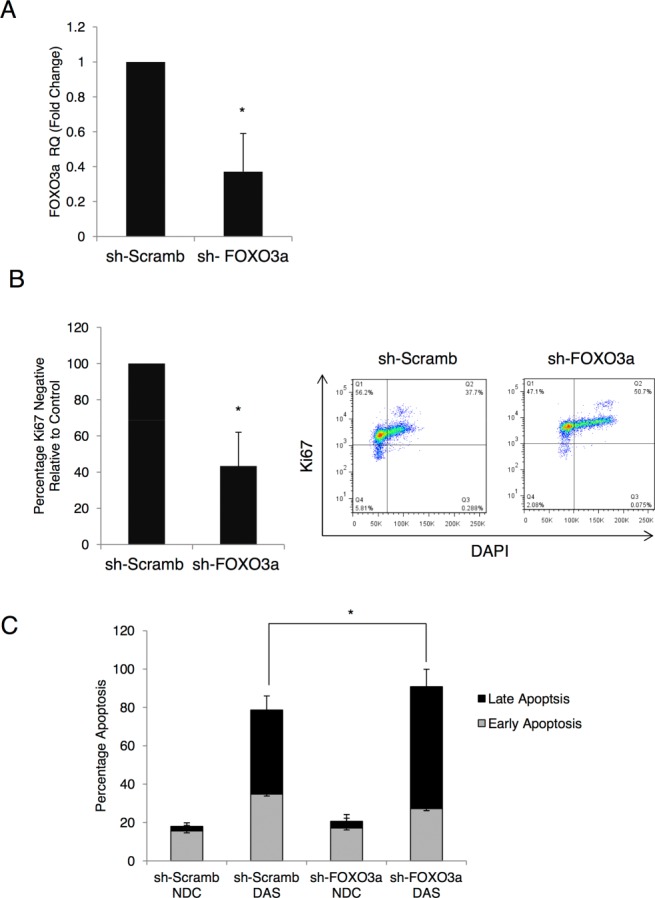
Chronic myeloid leukemia (CML) is initiated and maintained by the tyrosine kinase BCR-ABL which activates a number of signal transduction pathways, including PI3K/AKT signaling and consequently inactivates FOXO transcription factors. ABL-specific tyrosine kinase inhibitors (TKIs) induce minimal apoptosis in CML progenitor cells, yet exert potent antiproliferative effects, through as yet poorly understood mechanisms. Here, we demonstrate that in CD34+ CML cells, FOXO1 and 3a are inactivated and relocalized to the cytoplasm by BCR-ABL activity. TKIs caused a decrease in phosphorylation of FOXOs, leading to their relocalization from cytoplasm (inactive) to nucleus (active), where they modulated the expression of key FOXO target genes, such as Cyclin D1, ATM, CDKN1C, and BCL6 and induced G1 arrest. Activation of FOXO1 and 3a and a decreased expression of their target gene Cyclin D1 were also observed after 6 days of in vivo treatment with dasatinib in a CML transgenic mouse model. The over-expression of FOXO3a in CML cells combined with TKIs to reduce proliferation, with similar results seen for inhibitors of PI3K/AKT/mTOR signaling. While stable expression of an active FOXO3a mutant induced a similar level of quiescence to TKIs alone, shRNA-mediated knockdown of FOXO3a drove CML cells into cell cycle and potentiated TKI-induced apoptosis. These data demonstrate that TKI-induced G1 arrest in CML cells is mediated through inhibition of the PI3K/AKT pathway and reactivation of FOXOs. This enhanced understanding of TKI activity and induced progenitor cell quiescence suggests that new therapeutic strategies for CML should focus on manipulation of this signaling network.
Keywords: BCR-ABL; CD34+ progenitor cells; Chronic myeloid leukemia; FOXO transcription factors; Quiescence; Tyrosine kinase inhibitors.
© 2014 The Authors. STEM CELLS Published by Wiley Periodicals, Inc. on behalf of AlphaMed Press.
Figures
References
-
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. - PubMed
-
- Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. - PubMed
-
- Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11:1029–1035. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
