

Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells
- PMID: 24810052
- PMCID: PMC4047902
- DOI: 10.1038/cddis.2014.182
Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells
Abstract
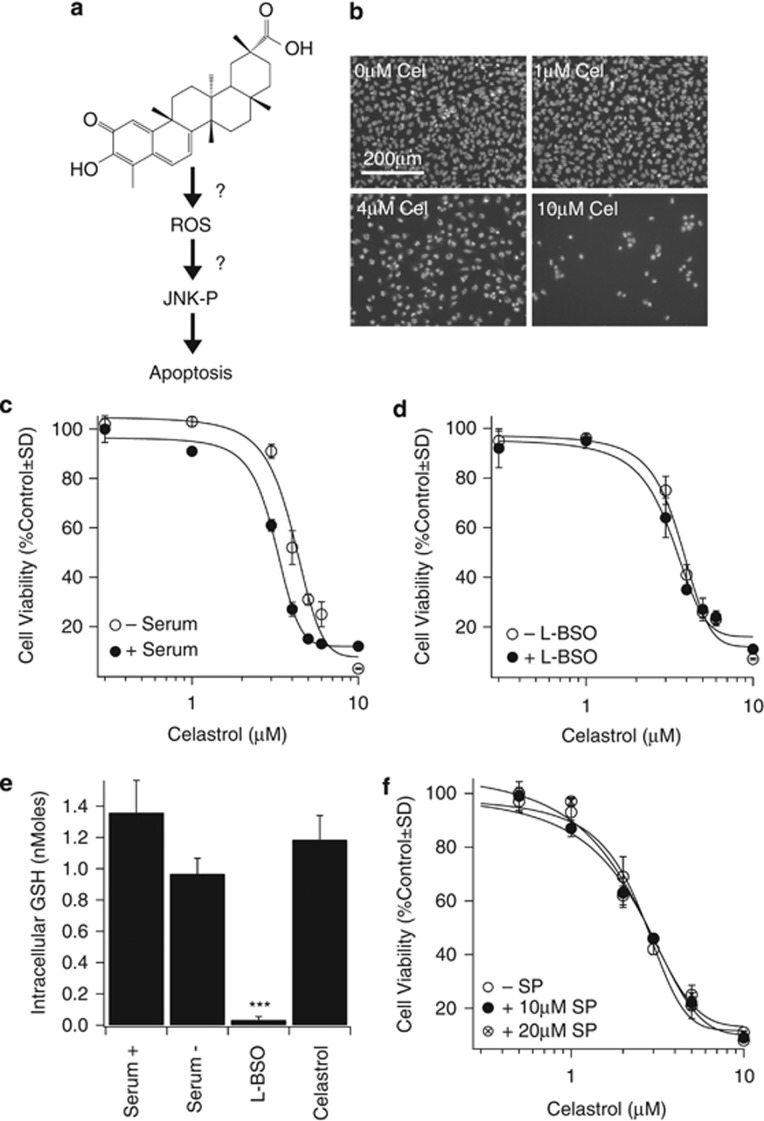
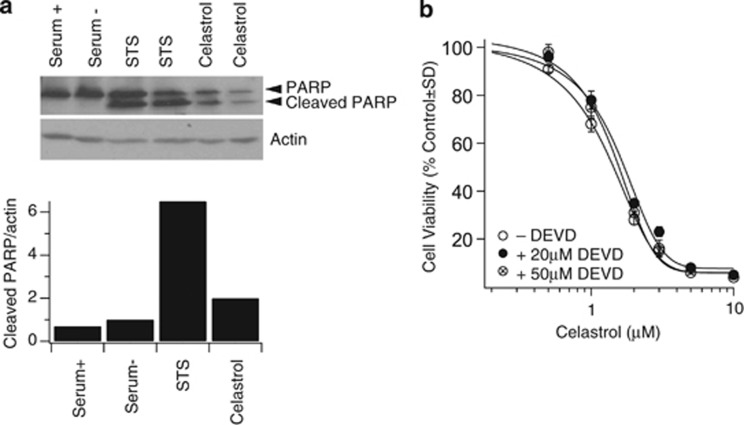
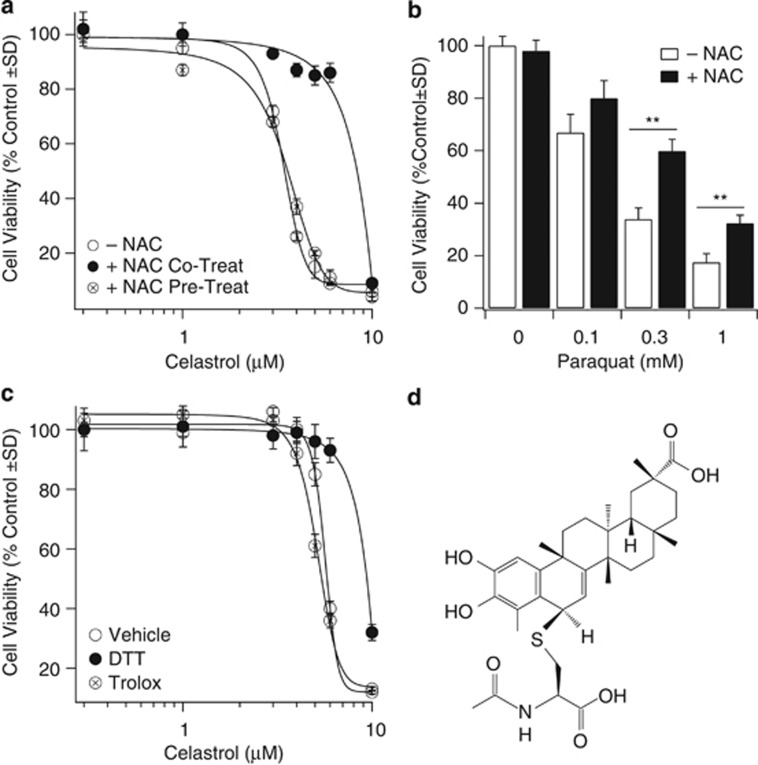
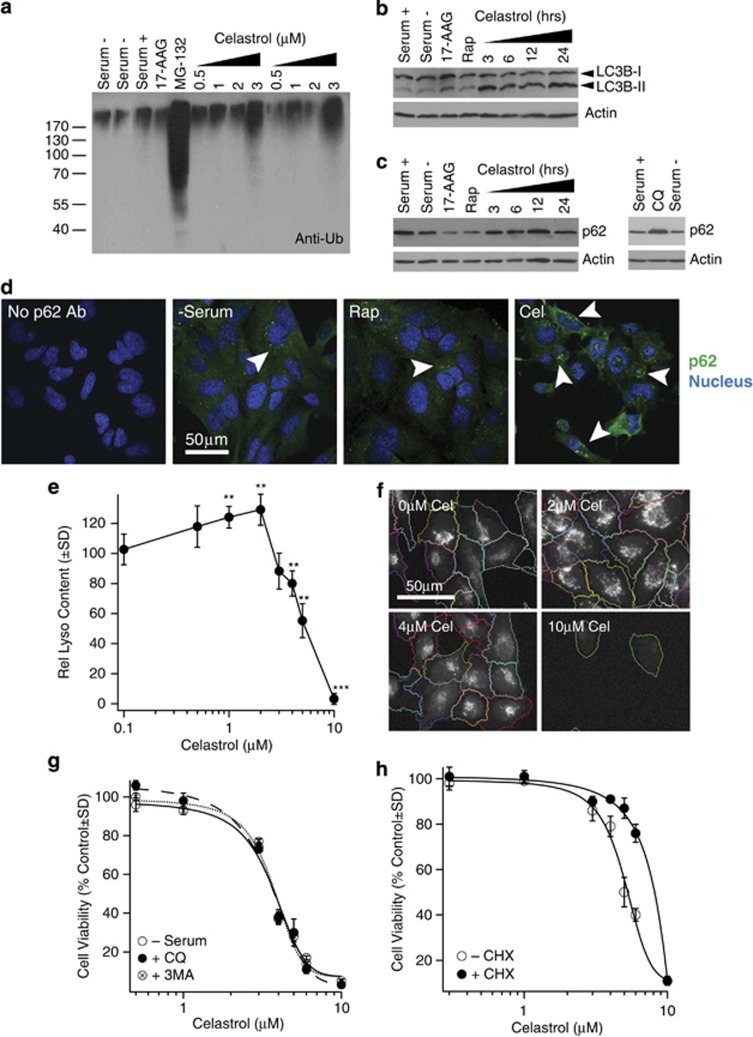
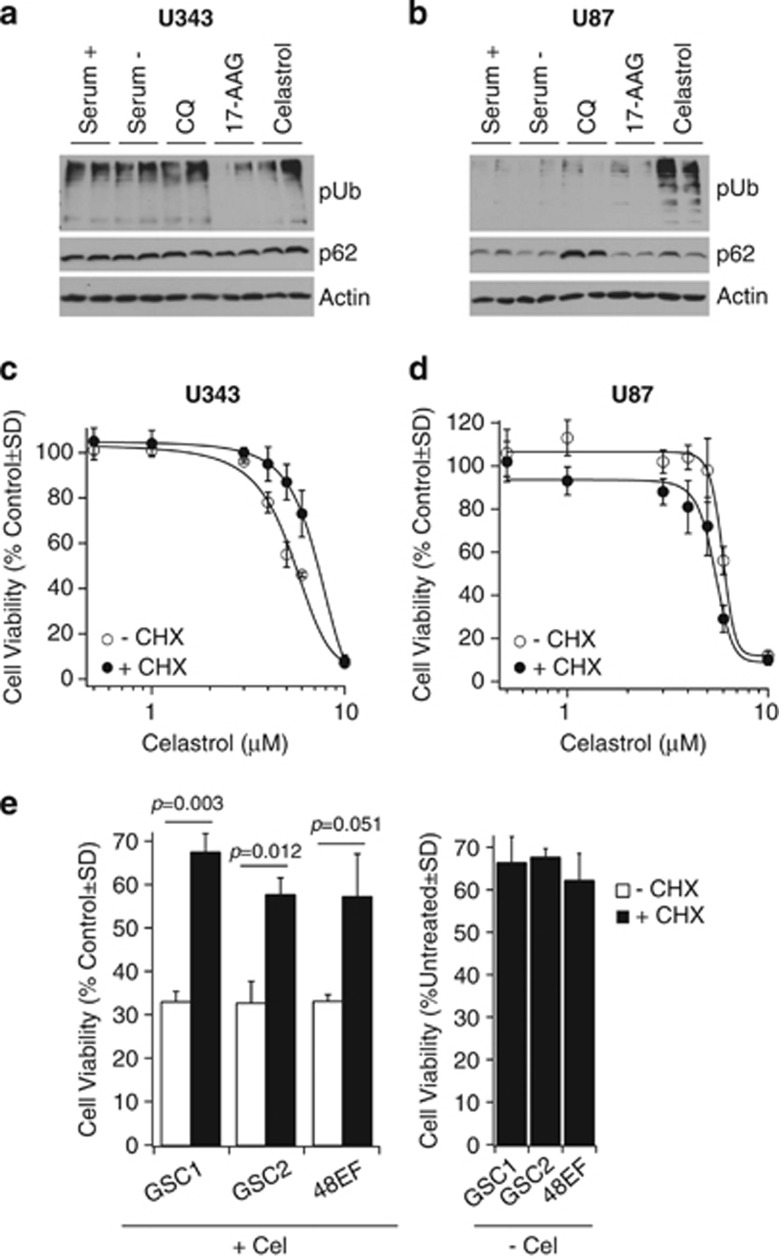
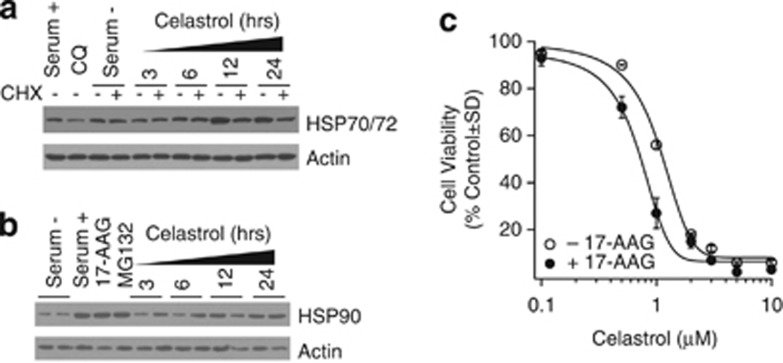
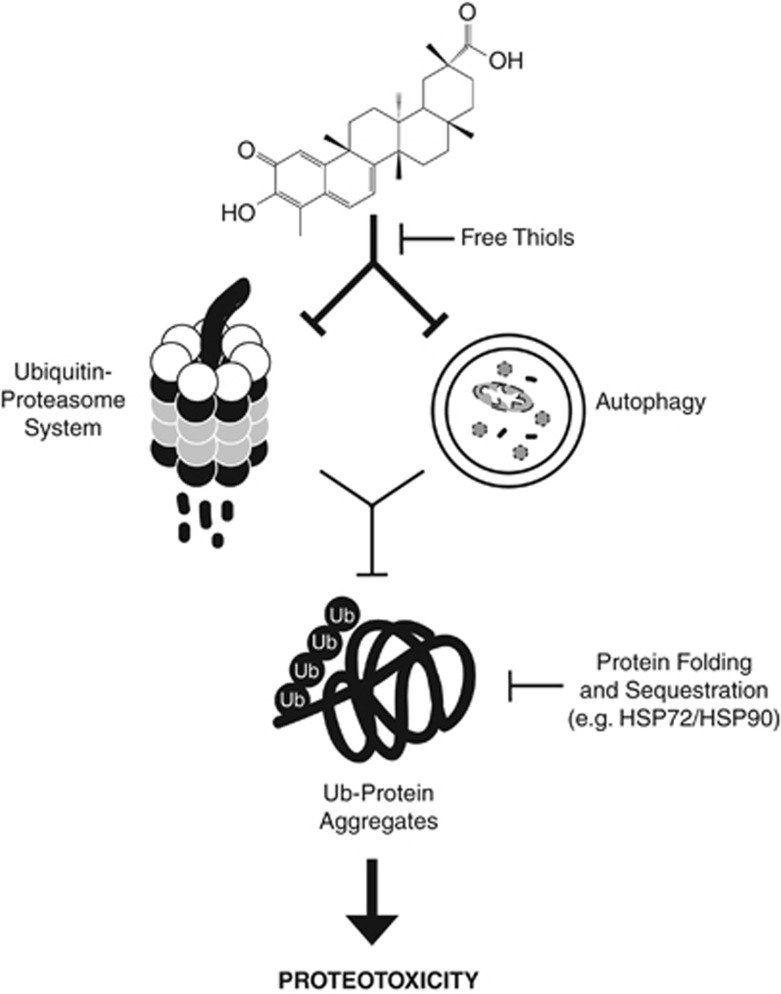
Glioblastoma multiforme is a devastating disease of the central nervous system and, at present, no effective therapeutic interventions have been identified. Celastrol, a natural occurring triterpene, exhibits potent anti-tumor activity against gliomas in xenograft mouse models. In this study, we describe the cell death mechanism employed by celastrol and identify secondary targets for effective combination therapy against glioblastoma cell survival. In contrast to the previously proposed reactive oxygen species (ROS)-dependent mechanism, cell death in human glioblastoma cells is shown here to be mediated by alternate signal transduction pathways involving, but not fully dependent on, poly(ADP-ribose) polymerase-1 and caspase-3. Our studies indicate that celastrol promotes proteotoxic stress, supported by two feedback mechanisms: (i) impairment of protein quality control as revealed by accumulation of polyubiquitinated aggregates and the canonical autophagy substrate, p62, and (ii) the induction of heat-shock proteins, HSP72 and HSP90. The Michael adduct of celastrol and N-acetylcysteine, 6-N-acetylcysteinyldihydrocelastrol, had no effect on p62, nor on HSP72 expression, confirming a thiol-dependent mechanism. Restriction of protein folding stress with cycloheximide was protective, while combination with autophagy inhibitors did not sensitize cells to celastrol-mediated cytotoxicity. Collectively, these findings imply that celastrol targets proteostasis by disrupting sulfyhydryl homeostasis, independently of ROS, in human glioblastoma cells. This study further emphasizes that targeting proteotoxic stress responses by inhibiting HSP90 with 17-N-Allylamino-17-demethoxygeldanamycin sensitizes human glioblastoma to celastrol treatment, thereby serving as a novel synergism to overcome drug resistance.
Figures
Similar articles
-
Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers.Cancer Biol Ther. 2011 Jan 15;11(2):263-76. doi: 10.4161/cbt.11.2.13959. Epub 2011 Jan 15. Cancer Biol Ther. 2011. PMID: 21088503 Free PMC article.
-
Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells.BMC Cancer. 2011 May 14;11:170. doi: 10.1186/1471-2407-11-170. BMC Cancer. 2011. PMID: 21569548 Free PMC article.
-
Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90.J Cell Physiol. 2012 May;227(5):2196-206. doi: 10.1002/jcp.22956. J Cell Physiol. 2012. PMID: 21866552
-
Recent Trends in anti-tumor mechanisms and molecular targets of celastrol.Int J Biol Sci. 2024 Oct 7;20(14):5510-5530. doi: 10.7150/ijbs.99592. eCollection 2024. Int J Biol Sci. 2024. PMID: 39494324 Free PMC article. Review.
-
Autophagy in the pharmacological activities of celastrol (Review).Exp Ther Med. 2023 Apr 20;25(6):268. doi: 10.3892/etm.2023.11967. eCollection 2023 Jun. Exp Ther Med. 2023. PMID: 37206564 Free PMC article. Review.
Cited by
-
The Interaction of the Metallo-Glycopeptide Anti-Tumour Drug Bleomycin with DNA.Int J Mol Sci. 2018 May 4;19(5):1372. doi: 10.3390/ijms19051372. Int J Mol Sci. 2018. PMID: 29734689 Free PMC article. Review.
-
A Mechanistic Overview of Triptolide and Celastrol, Natural Products from Tripterygium wilfordii Hook F.Front Pharmacol. 2018 Feb 14;9:104. doi: 10.3389/fphar.2018.00104. eCollection 2018. Front Pharmacol. 2018. PMID: 29491837 Free PMC article. Review.
-
Mevalonate-derived quinonemethide triterpenoid from in vitro roots of Peritassa laevigata and their localization in root tissue by MALDI imaging.Sci Rep. 2016 Mar 4;6:22627. doi: 10.1038/srep22627. Sci Rep. 2016. PMID: 26943243 Free PMC article.
-
Modulation of Inflammation-Related Lipid Mediator Pathways by Celastrol During Human Macrophage Polarization.J Inflamm Res. 2022 Jun 2;15:3285-3304. doi: 10.2147/JIR.S356964. eCollection 2022. J Inflamm Res. 2022. PMID: 35676971 Free PMC article.
-
Advances in HSP27 and HSP90-targeting strategies for glioblastoma.J Neurooncol. 2016 Apr;127(2):209-19. doi: 10.1007/s11060-016-2070-8. Epub 2016 Feb 2. J Neurooncol. 2016. PMID: 26842818 Review.
References
-
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. - PubMed
-
- Lima FR, Kahn SA, Soletti RC, Biasoli D, Alves T, da Fonseca AC, et al. Glioblastoma: therapeutic challenges, what lies ahead. Biochim Biophys Acta. 2012;1826:338–349. - PubMed
-
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. - PubMed
-
- Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013;331:139–146. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
