

Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8
- PMID: 24810276
- PMCID: PMC4014442
- DOI: 10.1371/journal.pgen.1004281
Genome sequencing and comparative genomics of the broad host-range pathogen Rhizoctonia solani AG8
Abstract
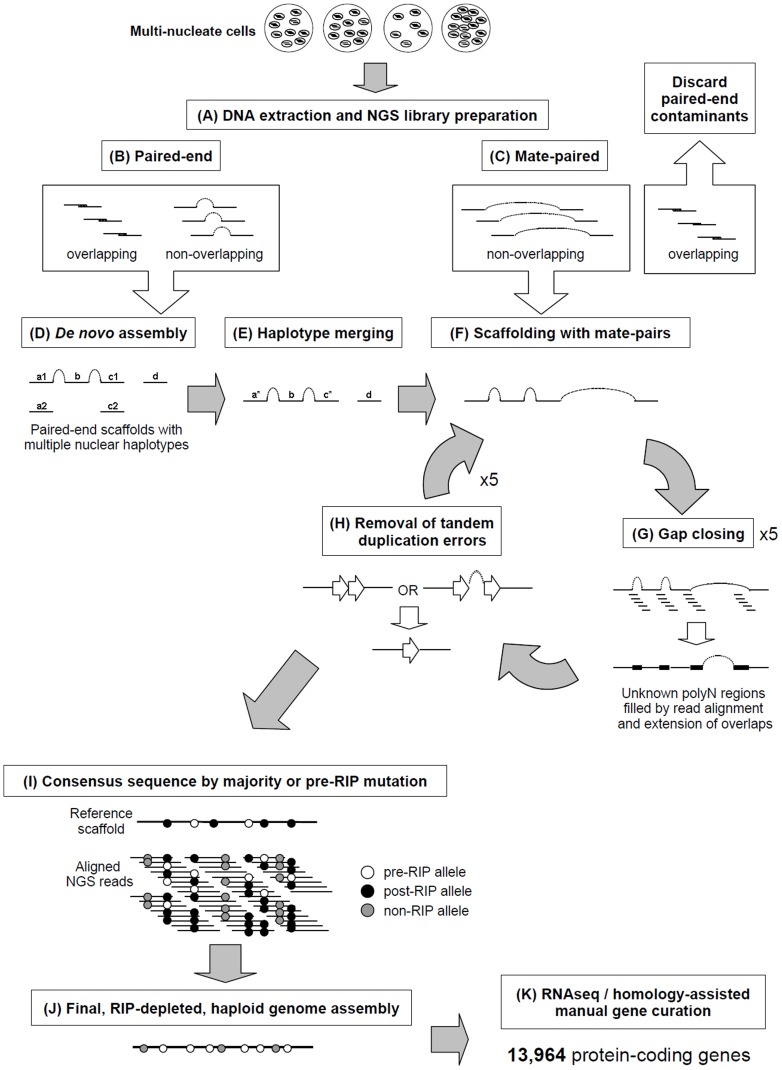
Rhizoctonia solani is a soil-borne basidiomycete fungus with a necrotrophic lifestyle which is classified into fourteen reproductively incompatible anastomosis groups (AGs). One of these, AG8, is a devastating pathogen causing bare patch of cereals, brassicas and legumes. R. solani is a multinucleate heterokaryon containing significant heterozygosity within a single cell. This complexity posed significant challenges for the assembly of its genome. We present a high quality genome assembly of R. solani AG8 and a manually curated set of 13,964 genes supported by RNA-seq. The AG8 genome assembly used novel methods to produce a haploid representation of its heterokaryotic state. The whole-genomes of AG8, the rice pathogen AG1-IA and the potato pathogen AG3 were observed to be syntenic and co-linear. Genes and functions putatively relevant to pathogenicity were highlighted by comparing AG8 to known pathogenicity genes, orthology databases spanning 197 phytopathogenic taxa and AG1-IA. We also observed SNP-level "hypermutation" of CpG dinucleotides to TpG between AG8 nuclei, with similarities to repeat-induced point mutation (RIP). Interestingly, gene-coding regions were widely affected along with repetitive DNA, which has not been previously observed for RIP in mononuclear fungi of the Pezizomycotina. The rate of heterozygous SNP mutations within this single isolate of AG8 was observed to be higher than SNP mutation rates observed across populations of most fungal species compared. Comparative analyses were combined to predict biological processes relevant to AG8 and 308 proteins with effector-like characteristics, forming a valuable resource for further study of this pathosystem. Predicted effector-like proteins had elevated levels of non-synonymous point mutations relative to synonymous mutations (dN/dS), suggesting that they may be under diversifying selection pressures. In addition, the distant relationship to sequenced necrotrophs of the Ascomycota suggests the R. solani genome sequence may prove to be a useful resource in future comparative analysis of plant pathogens.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





Similar articles
-
Development of a Rhizoctonia solani AG1-IB Specific Gene Model Enables Comparative Genome Analyses between Phytopathogenic R. solani AG1-IA, AG1-IB, AG3 and AG8 Isolates.PLoS One. 2015 Dec 21;10(12):e0144769. doi: 10.1371/journal.pone.0144769. eCollection 2015. PLoS One. 2015. PMID: 26690577 Free PMC article.
-
Comparative Genomics: Insights on the Pathogenicity and Lifestyle of Rhizoctonia solani.Int J Mol Sci. 2021 Feb 22;22(4):2183. doi: 10.3390/ijms22042183. Int J Mol Sci. 2021. PMID: 33671736 Free PMC article.
-
Transcriptome analysis reveals molecular mechanisms of sclerotial development in the rice sheath blight pathogen Rhizoctonia solani AG1-IA.Funct Integr Genomics. 2019 Sep;19(5):743-758. doi: 10.1007/s10142-019-00677-0. Epub 2019 May 3. Funct Integr Genomics. 2019. PMID: 31054140
-
Molecular and genetic aspects of controlling the soilborne necrotrophic pathogens Rhizoctonia and Pythium.Plant Sci. 2014 Nov;228:61-70. doi: 10.1016/j.plantsci.2014.02.001. Epub 2014 Feb 12. Plant Sci. 2014. PMID: 25438786 Review.
-
Tobacco leaf spot and root rot caused by Rhizoctonia solani Kühn.Mol Plant Pathol. 2011 Apr;12(3):209-16. doi: 10.1111/j.1364-3703.2010.00664.x. Epub 2010 Oct 1. Mol Plant Pathol. 2011. PMID: 21355993 Free PMC article. Review.
Cited by
-
Draft genome sequence of fastidious pathogen Ceratobasidium theobromae, which causes vascular-streak dieback in Theobroma cacao.Fungal Biol Biotechnol. 2019 Sep 30;6:14. doi: 10.1186/s40694-019-0077-6. eCollection 2019. Fungal Biol Biotechnol. 2019. PMID: 31583107 Free PMC article.
-
Intraspecific comparative genomics of isolates of the Norway spruce pathogen (Heterobasidion parviporum) and identification of its potential virulence factors.BMC Genomics. 2018 Mar 27;19(1):220. doi: 10.1186/s12864-018-4610-4. BMC Genomics. 2018. PMID: 29580224 Free PMC article.
-
Comparative Mitogenomic Analysis and the Evolution of Rhizoctonia solani Anastomosis Groups.Front Microbiol. 2021 Sep 20;12:707281. doi: 10.3389/fmicb.2021.707281. eCollection 2021. Front Microbiol. 2021. PMID: 34616376 Free PMC article.
-
Molecular Detection Assays for Rapid Field-Detection of Rice Sheath Blight.Front Plant Sci. 2021 Jan 11;11:552916. doi: 10.3389/fpls.2020.552916. eCollection 2020. Front Plant Sci. 2021. PMID: 33505407 Free PMC article.
-
An In vitro Study of Bio-Control and Plant Growth Promotion Potential of Salicaceae Endophytes.Front Microbiol. 2017 Mar 13;8:386. doi: 10.3389/fmicb.2017.00386. eCollection 2017. Front Microbiol. 2017. PMID: 28348550 Free PMC article.
References
-
- Sneh B, Burpee L, Ogoshi A, editors(1991) Identification of Rhizoctonia species. St. Paul, Minnesota, USA: APS Press.
-
- Paulitz TC (2006) Low input no-till cereal production in the Pacific Northwest of the US: The challenges of root diseases. Eur J Plant Pathol 115: 271–281.
-
- Bell DK, Sumner DR (1982) Virulence of Rhizoctonia solani Ag-2 Type-1 and Type-2 and Ag-4 from Peanut Seed on Corn, Sorghum, Lupine, Snapbean, Peanut and Soybean. Phytopathology 72: 947–948.
-
- Sumner DR, Bell DK (1982) Crop-Rotation and Yield Loss in Corn in Soil Infested with Rhizoctonia solani Ag-2 and Ag-4. Phytopathology 72: 361–362.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
