

Detecting and characterizing circular RNAs
- PMID: 24811520
- PMCID: PMC4121655
- DOI: 10.1038/nbt.2890
Detecting and characterizing circular RNAs
Abstract
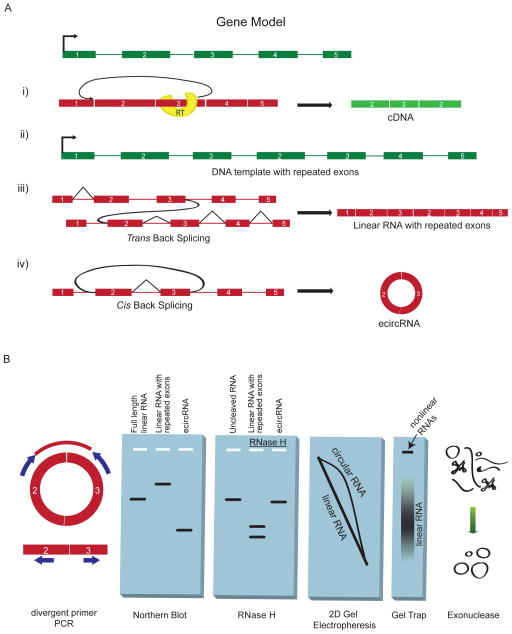
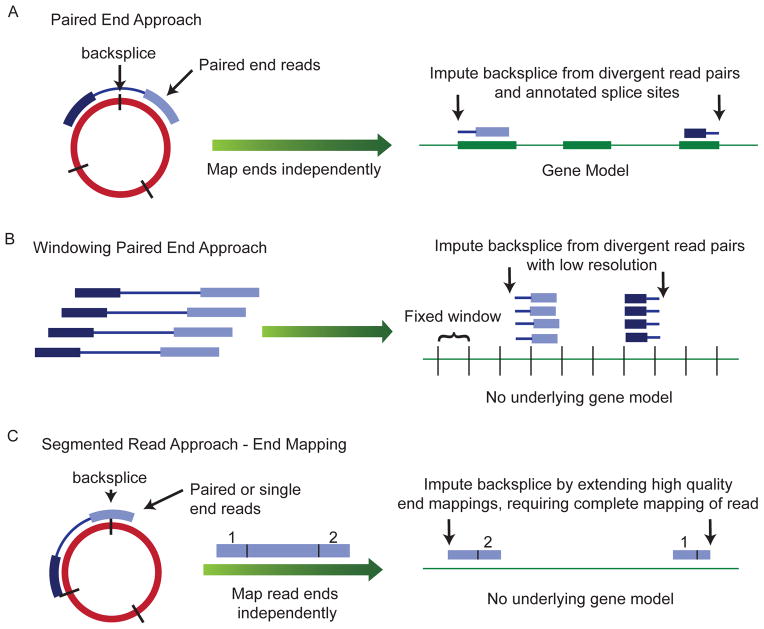
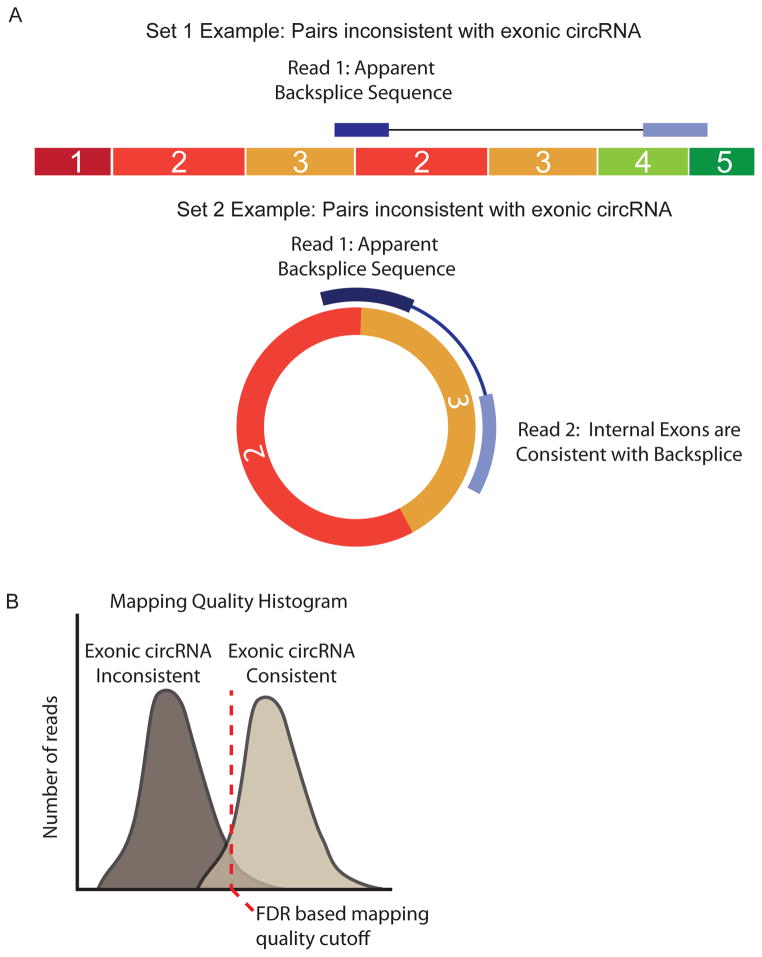
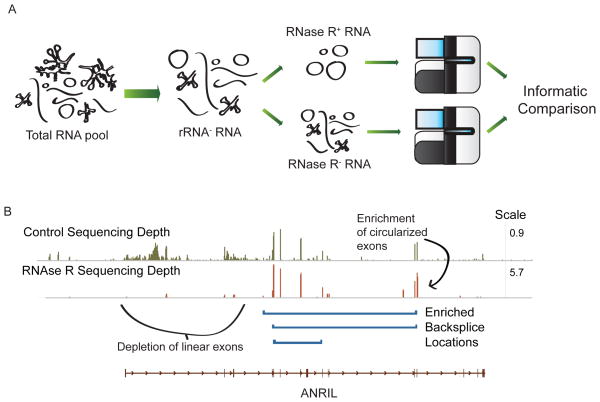
Circular RNA transcripts were first identified in the early 1990s but knowledge of these species has remained limited, as their study through traditional methods of RNA analysis has been difficult. Now, novel bioinformatic approaches coupled with biochemical enrichment strategies and deep sequencing have allowed comprehensive studies of circular RNA species. Recent studies have revealed thousands of endogenous circular RNAs in mammalian cells, some of which are highly abundant and evolutionarily conserved. Evidence is emerging that some circRNAs might regulate microRNA (miRNA) function, and roles in transcriptional control have also been suggested. Therefore, study of this class of noncoding RNAs has potential implications for therapeutic and research applications. We believe the key future challenge for the field will be to understand the regulation and function of these unusual molecules.
Figures
References
-
- Nigro JM, et al. Scrambled exons. Cell. 1991;64:607–613. - PubMed
-
- Cocquerelle C, Mascrez B, Hétuin D, Bailleul B. Mis-splicing yields circular RNA molecules. The FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 1993;7:155–160. - PubMed
-
- Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H. The hepatitis delta (delta) virus possesses a circular RNA. Nature. 1986;323:558–560. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
