

Efficient RNA isoform identification and quantification from RNA-Seq data with network flows
- PMID: 24813214
- PMCID: PMC4147886
- DOI: 10.1093/bioinformatics/btu317
Efficient RNA isoform identification and quantification from RNA-Seq data with network flows
Abstract
Motivation: Several state-of-the-art methods for isoform identification and quantification are based on [Formula: see text]-regularized regression, such as the Lasso. However, explicitly listing the-possibly exponentially-large set of candidate transcripts is intractable for genes with many exons. For this reason, existing approaches using the [Formula: see text]-penalty are either restricted to genes with few exons or only run the regression algorithm on a small set of preselected isoforms.
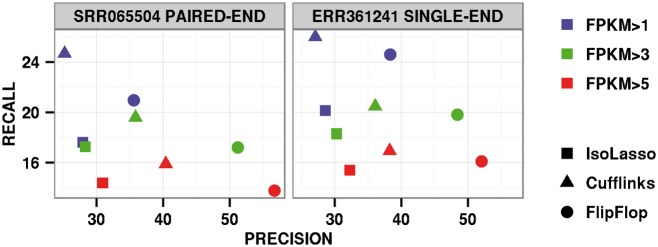
Results: We introduce a new technique called FlipFlop, which can efficiently tackle the sparse estimation problem on the full set of candidate isoforms by using network flow optimization. Our technique removes the need of a preselection step, leading to better isoform identification while keeping a low computational cost. Experiments with synthetic and real RNA-Seq data confirm that our approach is more accurate than alternative methods and one of the fastest available.
Availability and implementation: Source code is freely available as an R package from the Bioconductor Web site (http://www.bioconductor.org/), and more information is available at http://cbio.ensmp.fr/flipflop.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2014. Published by Oxford University Press.
Figures
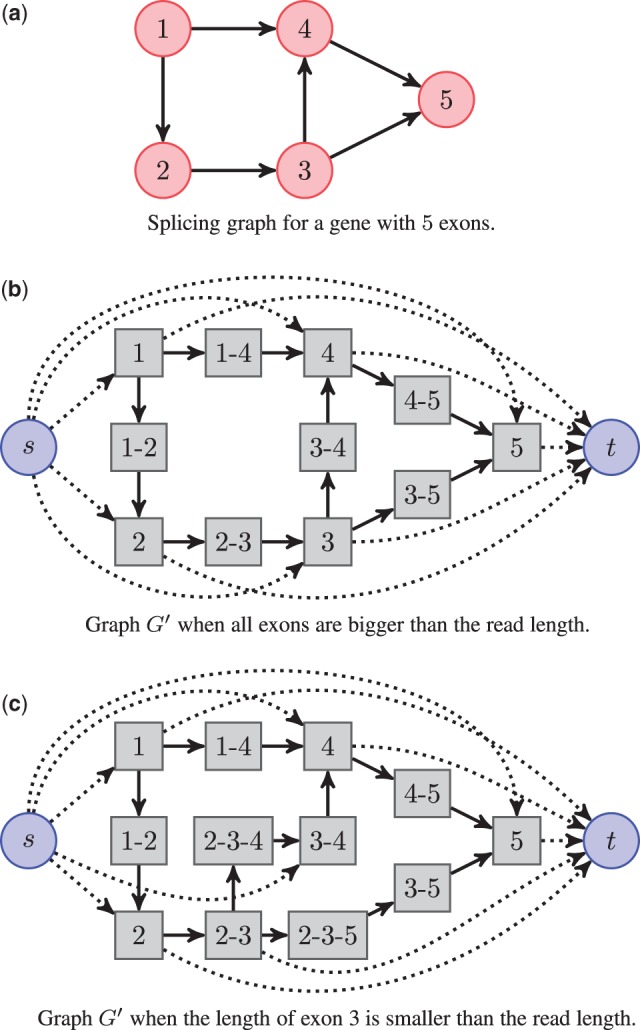
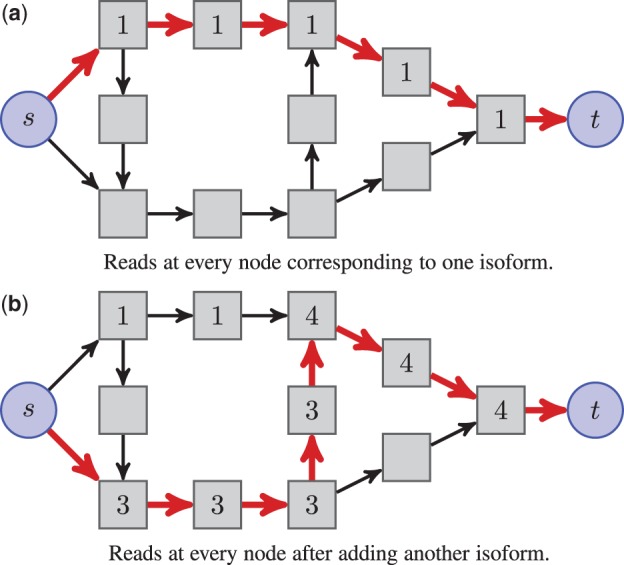
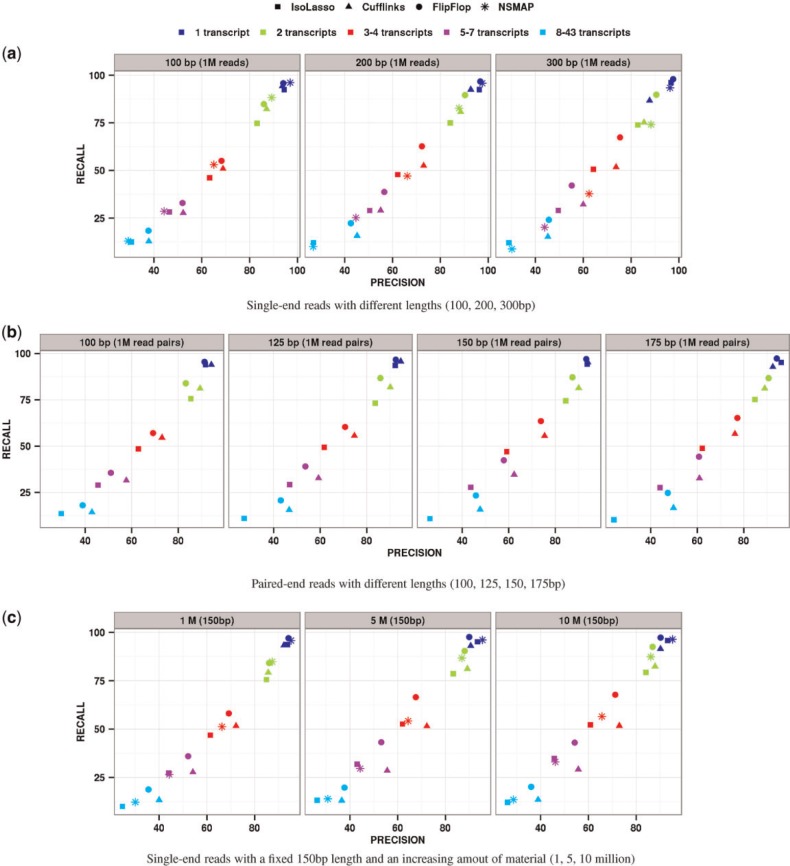
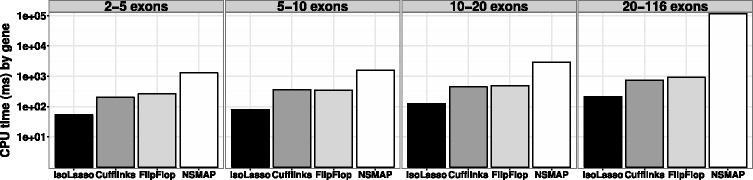
References
-
- Ahuja RK, et al. Network Flows: Theory, Algorithms, and Applications. Englewood Cliffs, New Jersey: Prentice Hall; 1993.
-
- Bertsekas DP. Network Optimization: Continuous and Discrete Models. Athena Scientific, Belmont, Massachusetts: Athena Scientific; 1998.
-
- Goldberg AV. An efficient implementation of a scaling minimum-cost flow algorithm. J. Algorithm. 1997;22:1–29.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
