

Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction
- PMID: 24813611
- PMCID: PMC4625837
- DOI: 10.1016/j.cell.2014.03.026
Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction
Abstract
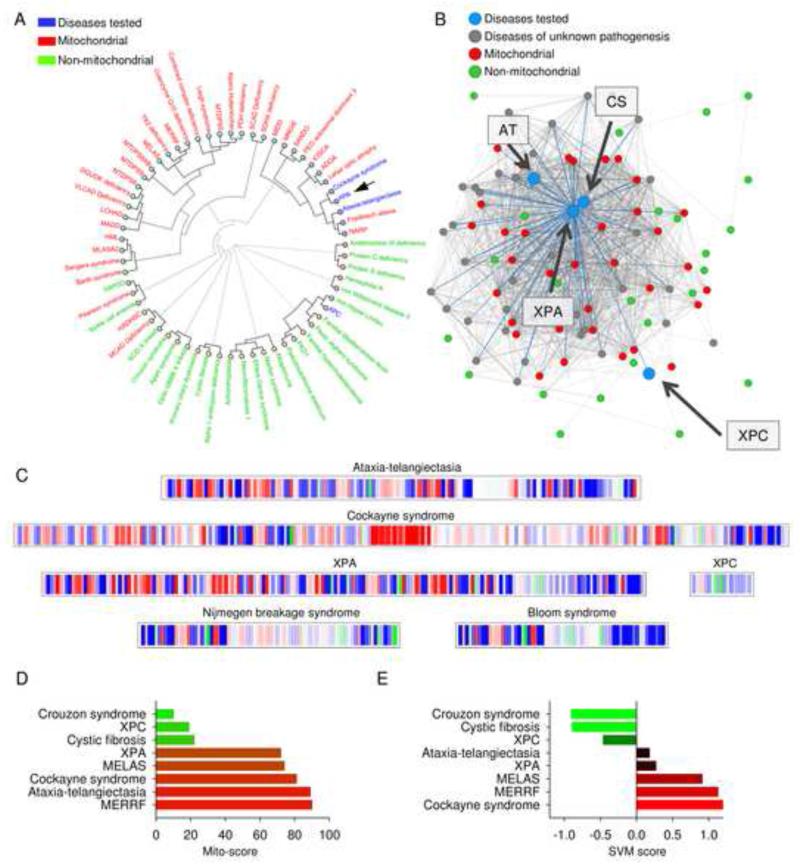
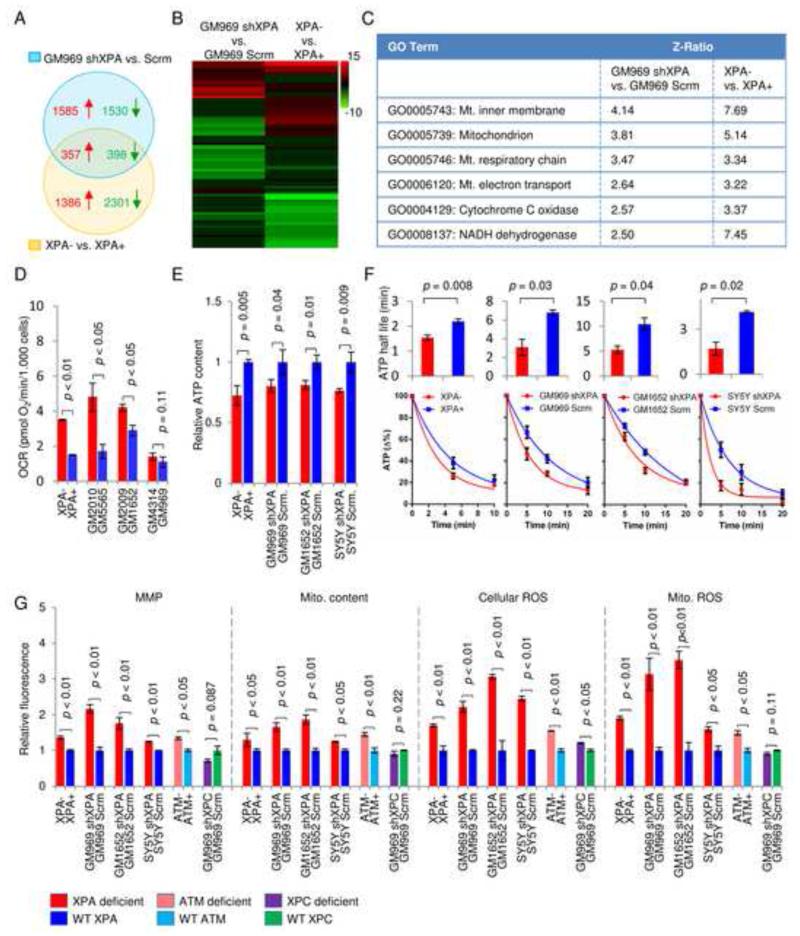
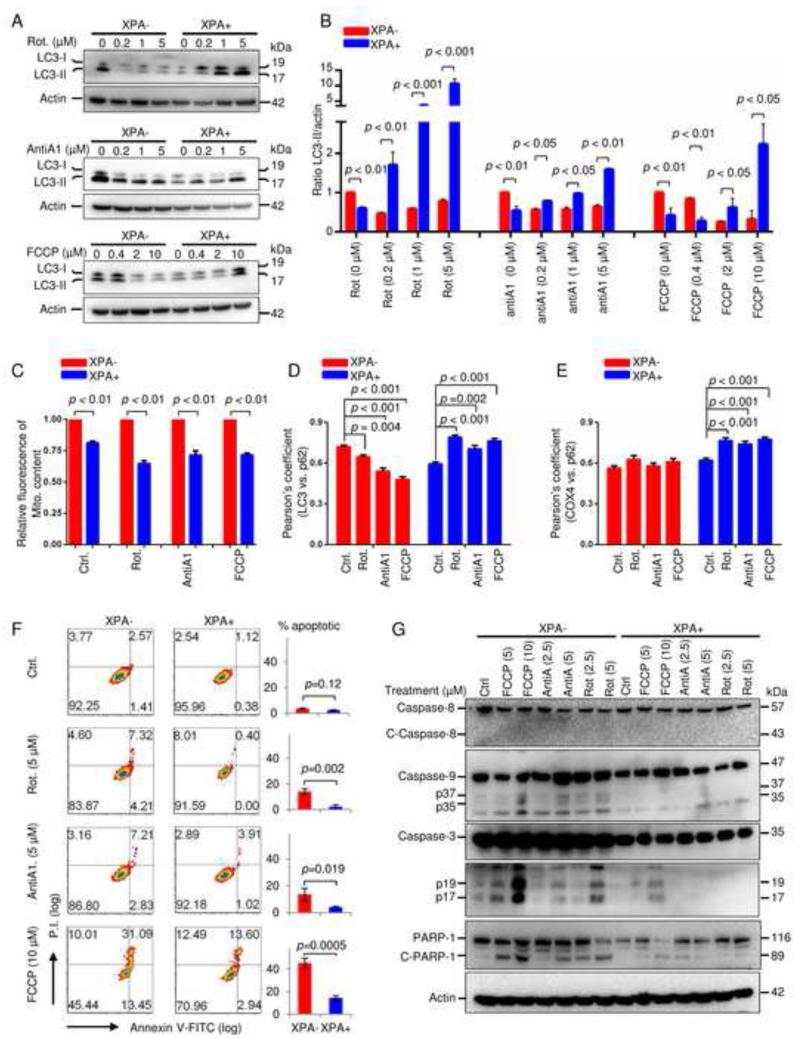
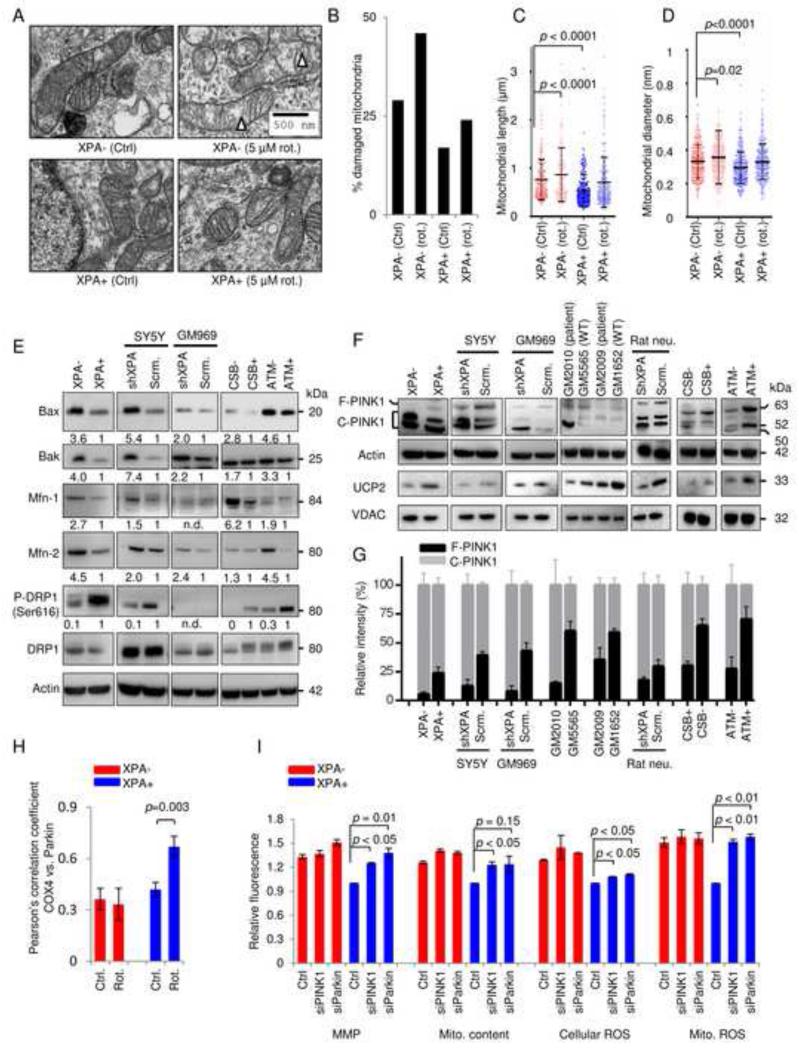
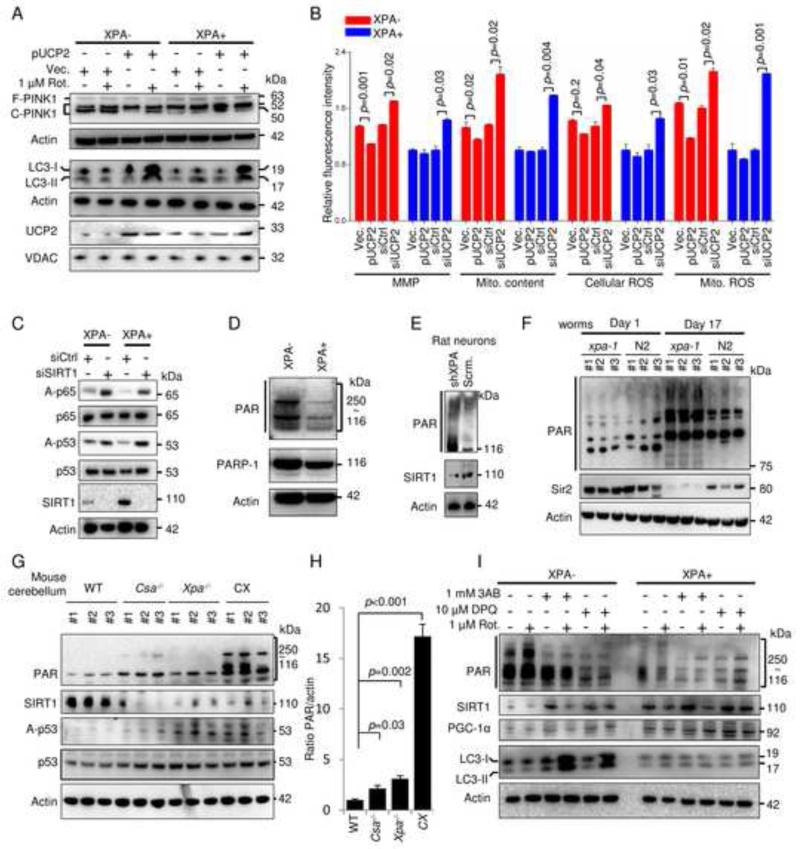
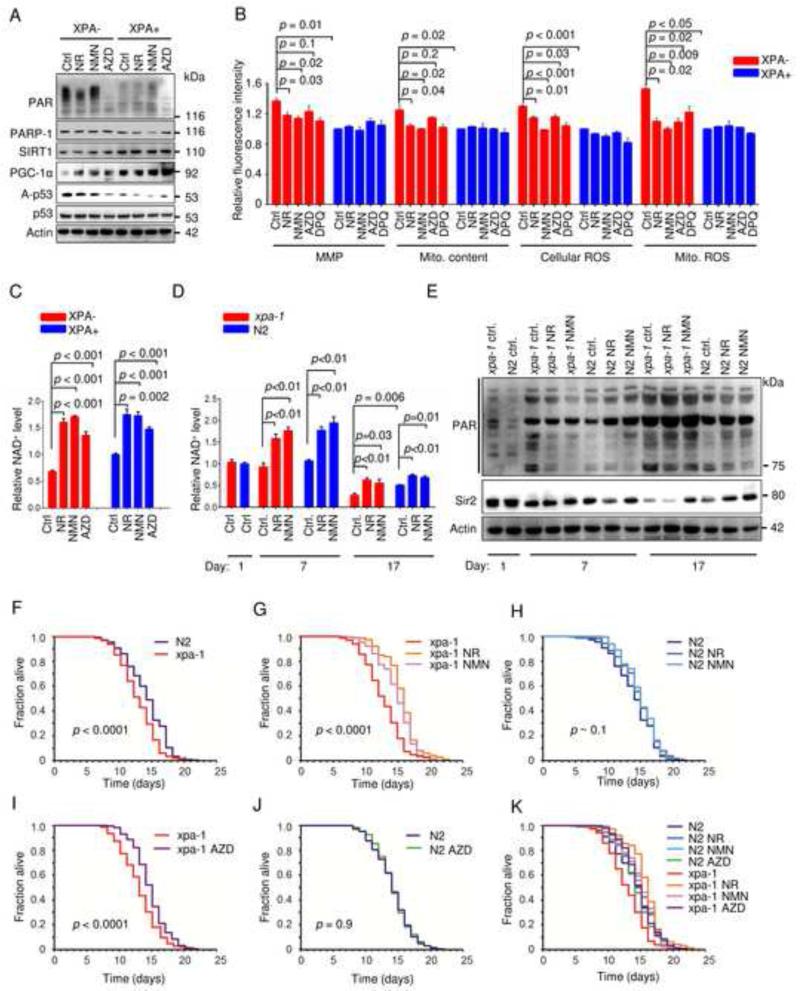
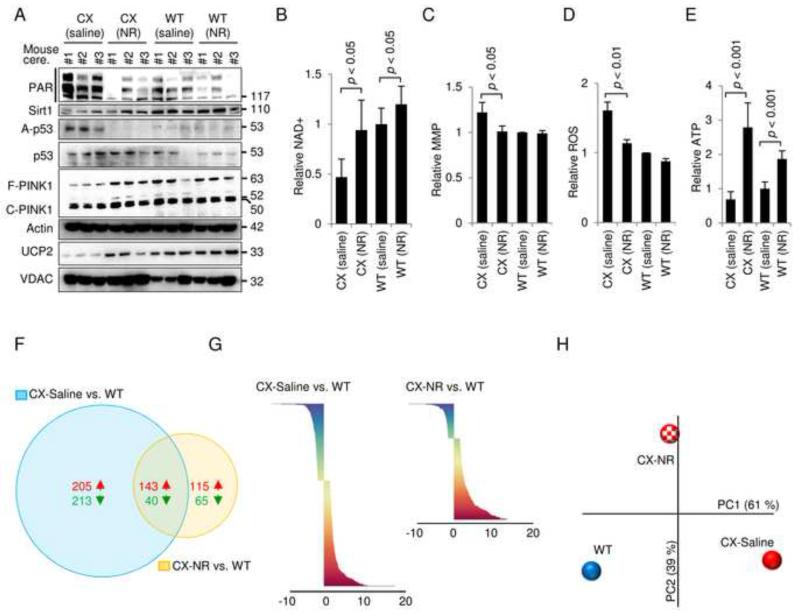
Mitochondrial dysfunction is a common feature in neurodegeneration and aging. We identify mitochondrial dysfunction in xeroderma pigmentosum group A (XPA), a nucleotide excision DNA repair disorder with severe neurodegeneration, in silico and in vivo. XPA-deficient cells show defective mitophagy with excessive cleavage of PINK1 and increased mitochondrial membrane potential. The mitochondrial abnormalities appear to be caused by decreased activation of the NAD(+)-SIRT1-PGC-1α axis triggered by hyperactivation of the DNA damage sensor PARP-1. This phenotype is rescued by PARP-1 inhibition or by supplementation with NAD(+) precursors that also rescue the lifespan defect in xpa-1 nematodes. Importantly, this pathogenesis appears common to ataxia-telangiectasia and Cockayne syndrome, two other DNA repair disorders with neurodegeneration, but absent in XPC, a DNA repair disorder without neurodegeneration. Our findings reveal a nuclear-mitochondrial crosstalk that is critical for the maintenance of mitochondrial health.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Contribution of defective mitophagy to the neurodegeneration in DNA repair-deficient disorders.Autophagy. 2014 Aug;10(8):1468-9. doi: 10.4161/auto.29321. Epub 2014 Jun 12. Autophagy. 2014. PMID: 24991831 Free PMC article.
-
Neurodegeneration in accelerated aging.Dan Med J. 2016 Nov;63(11):B5308. Dan Med J. 2016. PMID: 27808039 Review.
-
Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair.J Biol Chem. 2012 Nov 16;287(47):39824-33. doi: 10.1074/jbc.M112.393504. Epub 2012 Oct 4. J Biol Chem. 2012. PMID: 23038248 Free PMC article.
-
A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome.Cell Metab. 2014 Nov 4;20(5):840-855. doi: 10.1016/j.cmet.2014.10.005. Epub 2014 Nov 4. Cell Metab. 2014. PMID: 25440059 Free PMC article.
-
Xeroderma Pigmentosa Group A (XPA), Nucleotide Excision Repair and Regulation by ATR in Response to Ultraviolet Irradiation.Adv Exp Med Biol. 2017;996:41-54. doi: 10.1007/978-3-319-56017-5_4. Adv Exp Med Biol. 2017. PMID: 29124689 Free PMC article. Review.
Cited by
-
Roles of microglial mitophagy in neurological disorders.Front Aging Neurosci. 2022 Aug 10;14:979869. doi: 10.3389/fnagi.2022.979869. eCollection 2022. Front Aging Neurosci. 2022. PMID: 36034136 Free PMC article. Review.
-
Life-Cycle-Dependent Toxicities of Mono- and Bifunctional Alkylating Agents in the 3R-Compliant Model Organism C. elegans.Cells. 2023 Nov 29;12(23):2728. doi: 10.3390/cells12232728. Cells. 2023. PMID: 38067156 Free PMC article.
-
Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis.Redox Biol. 2020 Oct;37:101614. doi: 10.1016/j.redox.2020.101614. Epub 2020 Jun 24. Redox Biol. 2020. PMID: 32863187 Free PMC article. Review.
-
Mitochondria and chronic effects of cancer therapeutics: The clinical implications.J Thromb Thrombolysis. 2021 May;51(4):884-889. doi: 10.1007/s11239-020-02313-2. Epub 2020 Oct 20. J Thromb Thrombolysis. 2021. PMID: 33079380 Free PMC article. Review.
-
Autophagy requires poly(adp-ribosyl)ation-dependent AMPK nuclear export.Cell Death Differ. 2016 Dec;23(12):2007-2018. doi: 10.1038/cdd.2016.80. Epub 2016 Sep 30. Cell Death Differ. 2016. PMID: 27689873 Free PMC article.
References
-
- Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J, de Souza-Pinto NC, Scheibye-Knudsen M, Wilson DM, 3rd, Stevnsner T, et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:2334–2346. - PMC - PubMed
-
- Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson’s disease. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:184–191. - PMC - PubMed
-
- Arczewska KD, Tomazella GG, Lindvall JM, Kassahun H, Maglioni S, Torgovnick A, Henriksson J, Matilainen O, Marquis BJ, Nelson BC, et al. Active transcriptomic and proteomic reprogramming in the C. elegans nucleotide excision repair mutant xpa-1. Nucleic acids research. 2013;41:5368–5381. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
