

Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers
- PMID: 24813947
- PMCID: PMC4048654
- DOI: 10.1016/j.molcel.2014.04.006
Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers
Abstract
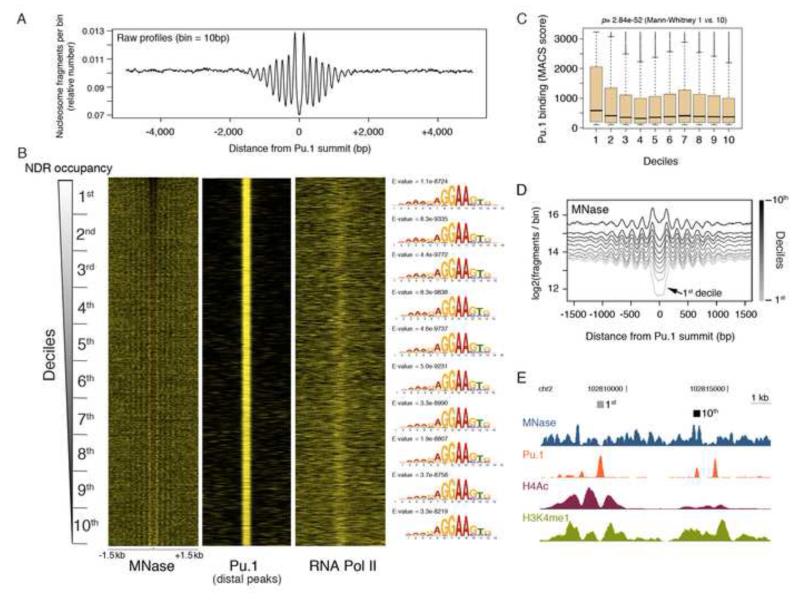
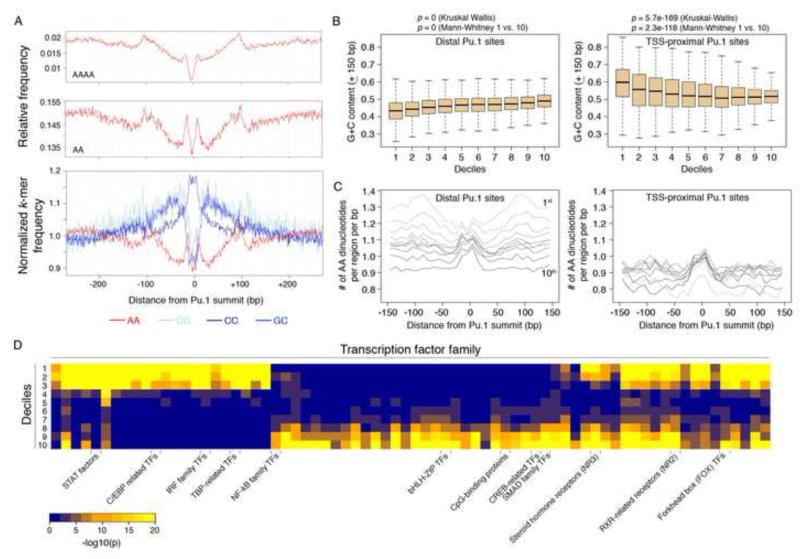
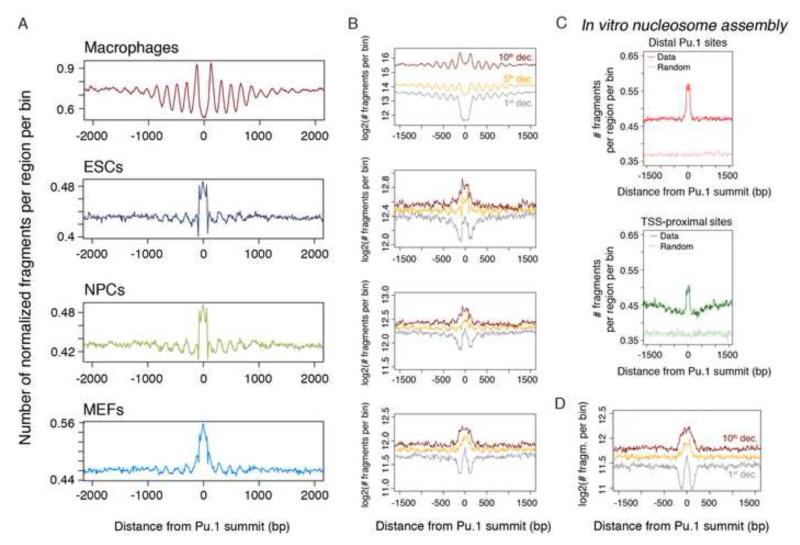
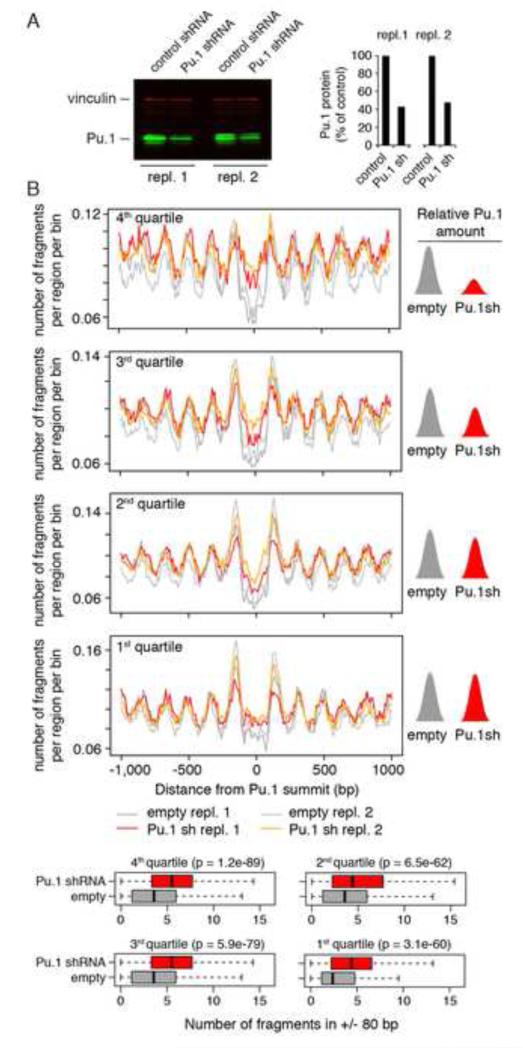
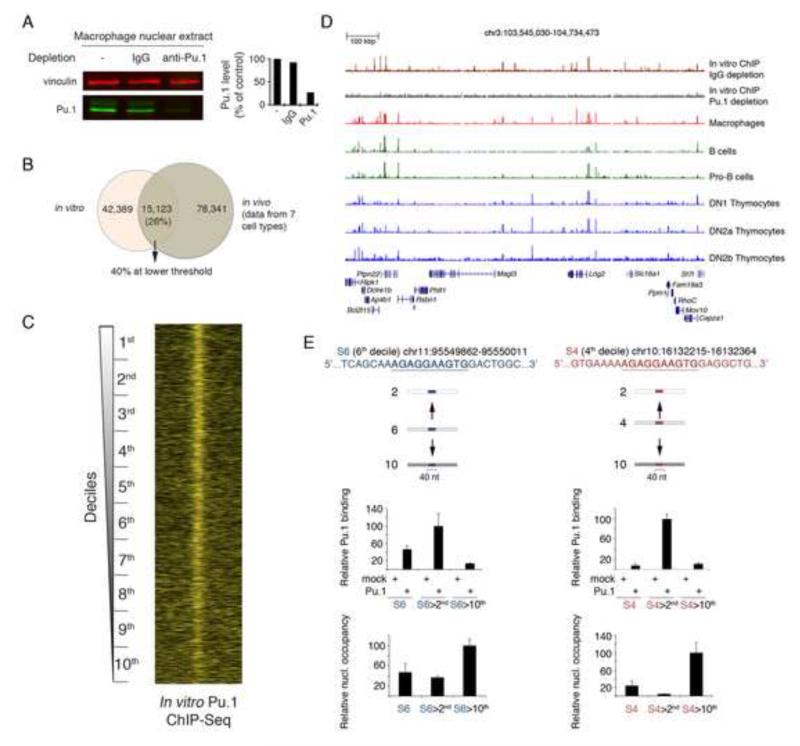
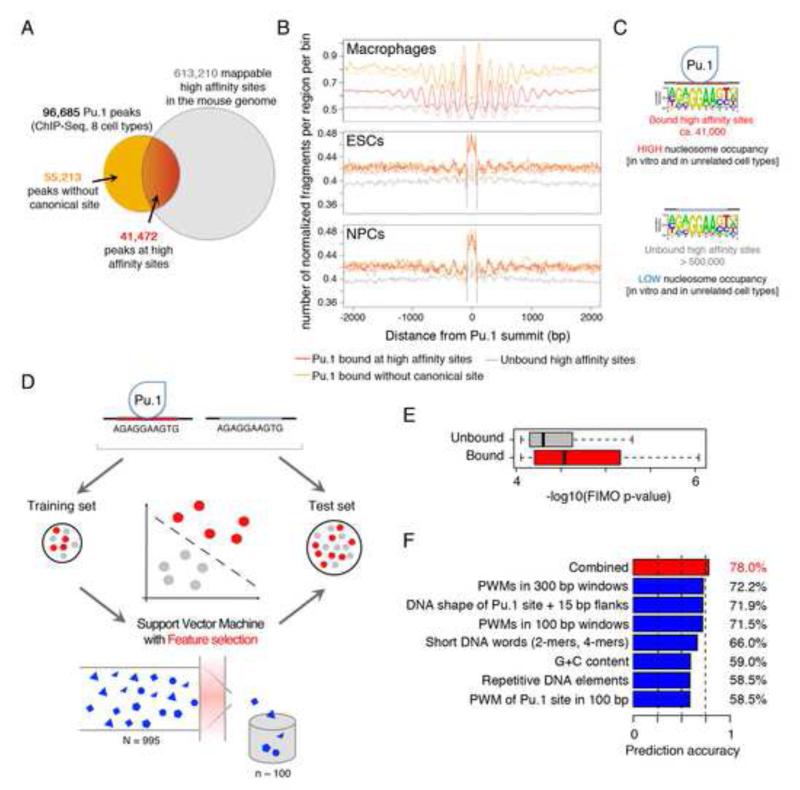
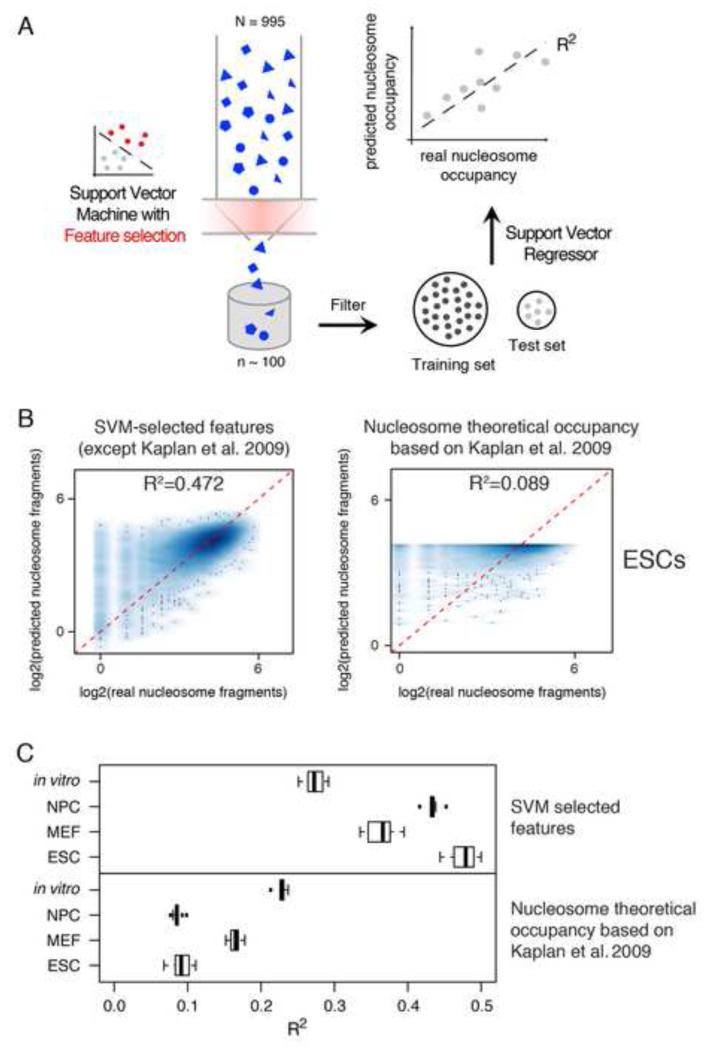
Transcription factors (TFs) preferentially bind sites contained in regions of computationally predicted high nucleosomal occupancy, suggesting that nucleosomes are gatekeepers of TF binding sites. However, because of their complexity mammalian genomes contain millions of randomly occurring, unbound TF consensus binding sites. We hypothesized that the information controlling nucleosome assembly may coincide with the information that enables TFs to bind cis-regulatory elements while ignoring randomly occurring sites. Hence, nucleosomes would selectively mask genomic sites that can be contacted by TFs and thus be potentially functional. The hematopoietic pioneer TF Pu.1 maintained nucleosome depletion at macrophage-specific enhancers that displayed a broad range of nucleosome occupancy in other cell types and in reconstituted chromatin. We identified a minimal set of DNA sequence and shape features that accurately predicted both Pu.1 binding and nucleosome occupancy genome-wide. These data reveal a basic organizational principle of mammalian cis-regulatory elements whereby TF recruitment and nucleosome deposition are controlled by overlapping DNA sequence features.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Chang C-C, Lin C-J. LIBSVM: A library for support vector machines. ACM Trans Intell Syst Technol. 2011;2:1–27.
-
- Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–297.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous