

Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy
- PMID: 24815523
- PMCID: PMC4108519
- DOI: 10.1016/j.hrthm.2014.04.042
Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy
Abstract
Background: Mutations in SCN5A, which encodes the cardiac sodium channel NaV1.5, typically cause ventricular arrhythmia or conduction slowing. Recently, SCN5A mutations have been associated with heart failure combined with variable atrial and ventricular arrhythmia.
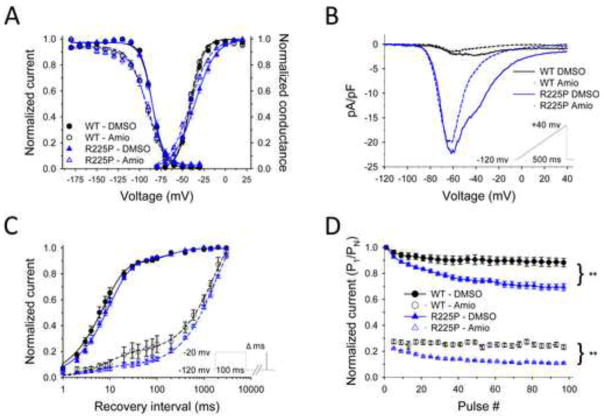
Objective: The purpose of this study was to determine the clinical, genetic, and functional features of an amiodarone-responsive multifocal ventricular ectopy-related cardiomyopathy associated with a novel mutation in a NaV1.5 voltage sensor domain.
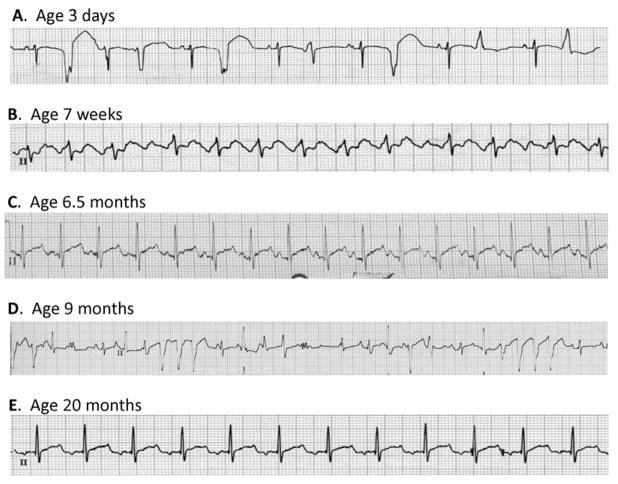
Methods: A novel, de novo SCN5A mutation (NaV1.5-R225P) was identified in a boy with prenatal arrhythmia and impaired cardiac contractility followed by postnatal multifocal ventricular ectopy suppressible by amiodarone. We investigated the functional consequences of NaV1.5-R225P expressed heterologously in tsA201 cells.
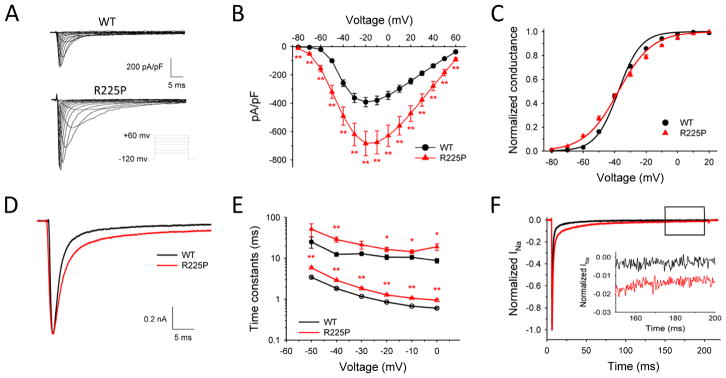
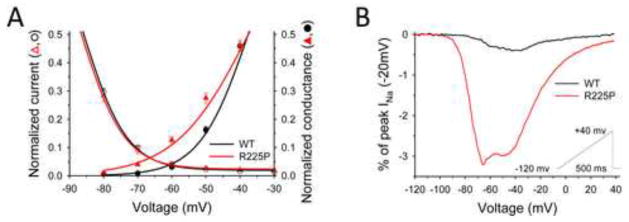
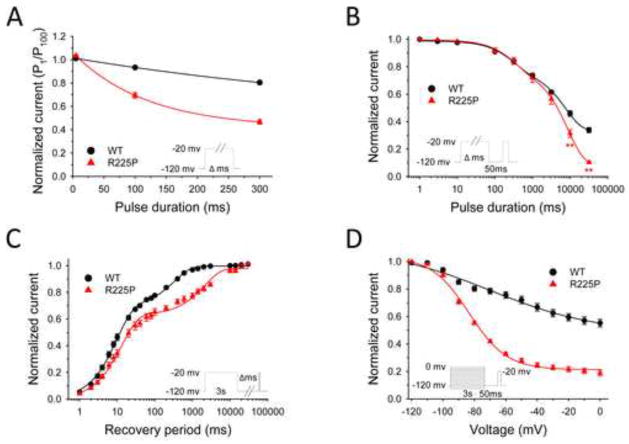
Results: Mutant channels exhibited significant abnormalities in both activation and inactivation leading to large, hyperpolarized window and ramp currents that predict aberrant sodium influx at potentials near the cardiomyocyte resting membrane potential. Mutant channels also exhibited significantly increased persistent (late) sodium current. This profile of channel dysfunction shares features with other SCN5A voltage sensor mutations associated with cardiomyopathy and overlapped that of congenital long QT syndrome. Amiodarone stabilized fast inactivation, suppressed persistent sodium current, and caused frequency-dependent inhibition of channel availability.
Conclusion: We determined the functional consequences and pharmacologic responses of a novel SCN5A mutation associated with an arrhythmia-associated cardiomyopathy. Comparisons with other cardiomyopathy-associated NaV1.5 voltage sensor mutations revealed a pattern of abnormal voltage dependence of activation as a shared biophysical mechanism of the syndrome.
Keywords: Amiodarone; Cardiomyopathy; Electrophysiology; SCN5A mutation.
Copyright © 2014 Heart Rhythm Society. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures

Comment in
-
SCN5A-related dilated cardiomyopathy: what do we know?Heart Rhythm. 2014 Aug;11(8):1454-5. doi: 10.1016/j.hrthm.2014.05.031. Epub 2014 May 29. Heart Rhythm. 2014. PMID: 24879950 No abstract available.
References
-
- George AL., Jr . Mechanisms in heritable sodium channel diseases. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. 6. Philadelphia: Elsevier; 2013.
-
- McNair WP, Ku L, Taylor MR, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. - PubMed
-
- Nguyen TP, Wang DW, Rhodes TH, George AL., Jr Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res. 2008;102:364–371. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
