

Faster fluorescence microscopy: advances in high speed biological imaging
- PMID: 24815857
- PMCID: PMC4096075
- DOI: 10.1016/j.cbpa.2014.04.008
Faster fluorescence microscopy: advances in high speed biological imaging
Abstract
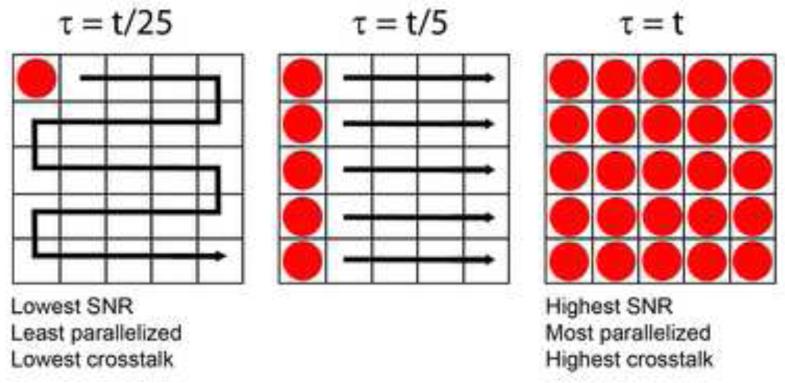
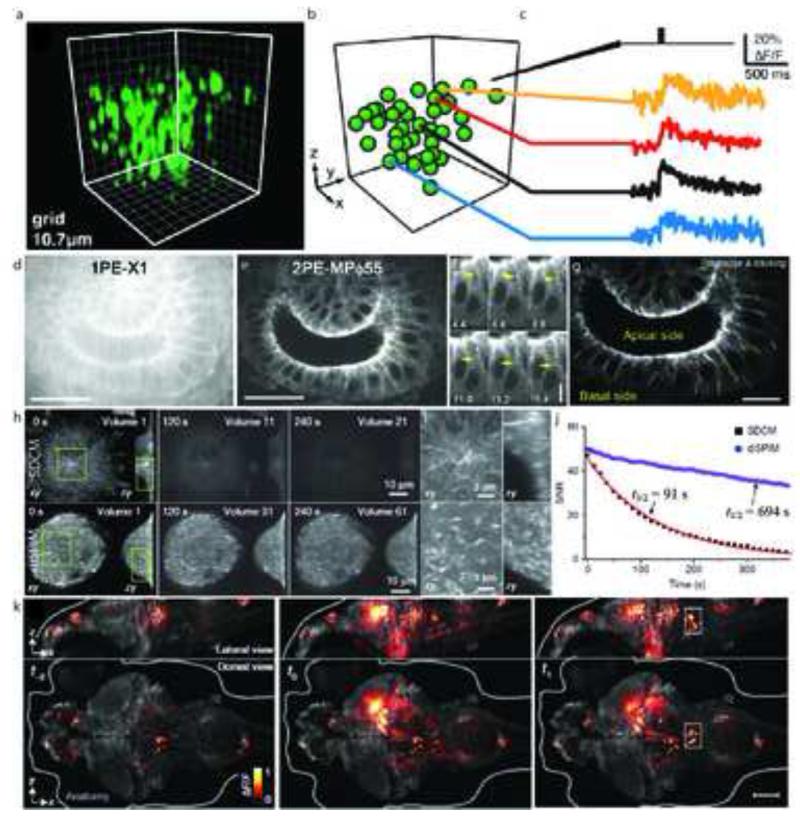
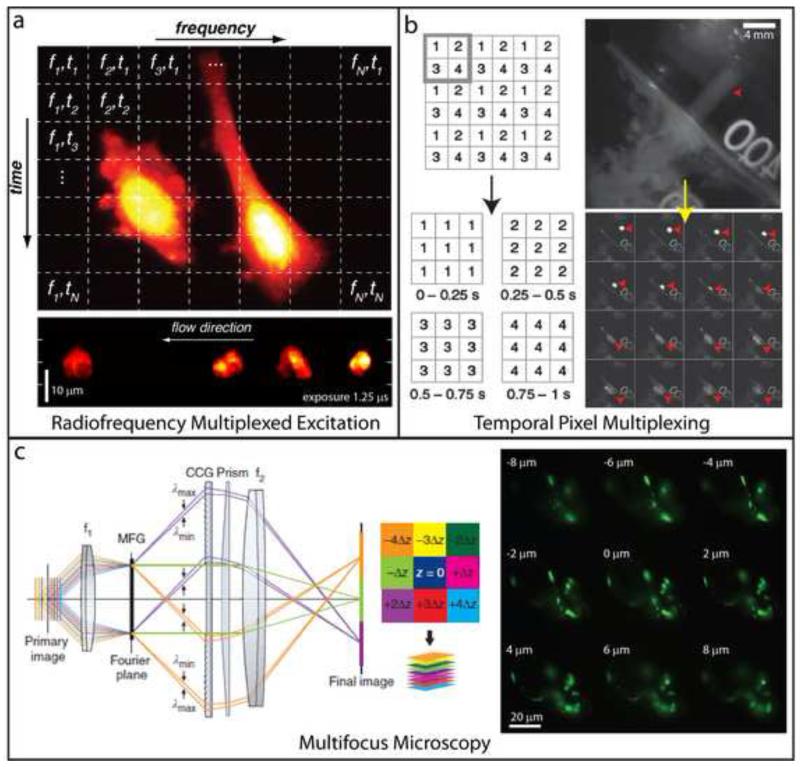
The past decade has seen explosive growth in new high speed imaging methods. These can broadly be classified as either point-scanning (which offer better depth penetration) or parallelized systems (which offer higher speed). We discuss each class generally, and cover specific advances in diffraction-limited microscopes (laser-scanning confocal, spinning-disk, and light-sheet) and superresolution microscopes (single-molecule imaging, stimulated emission-depletion, and structured illumination). A theme of our review is that there is no free lunch: each technique has strengths and weaknesses, and an advance in speed usually comes at the expense of either spatial resolution or depth penetration.
Published by Elsevier Ltd.
Figures

References
-
-
Katona G, Szalay G, Maak P, et al. Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nature Methods. 2012;9:201–208. Acousto-optic scanning technology permits high speed 3D imaging hundreds of times faster than galvanometric scanners, enabling 50 distinct points to be interrogated across nearly cm sized volumes every ms.
-
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
