

Structure of sulfamidase provides insight into the molecular pathology of mucopolysaccharidosis IIIA
- PMID: 24816101
- PMCID: PMC4014121
- DOI: 10.1107/S1399004714002739
Structure of sulfamidase provides insight into the molecular pathology of mucopolysaccharidosis IIIA
Abstract
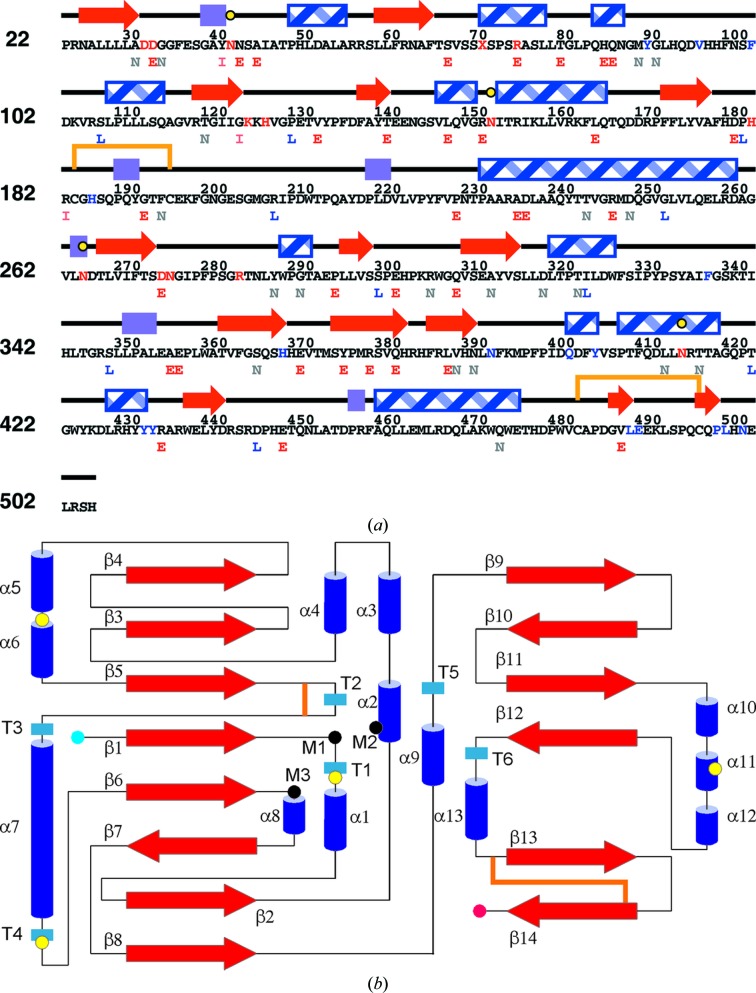
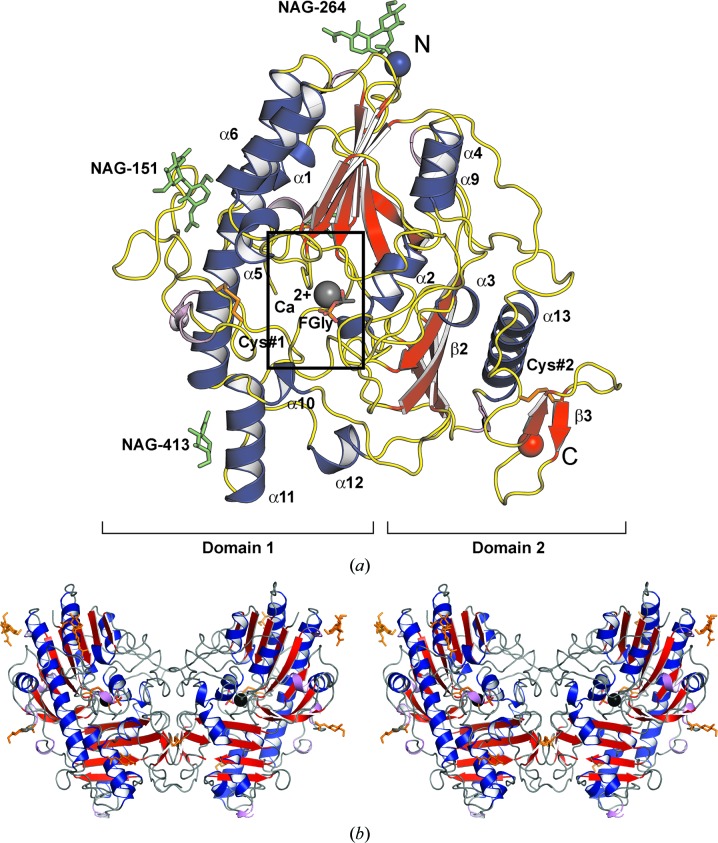
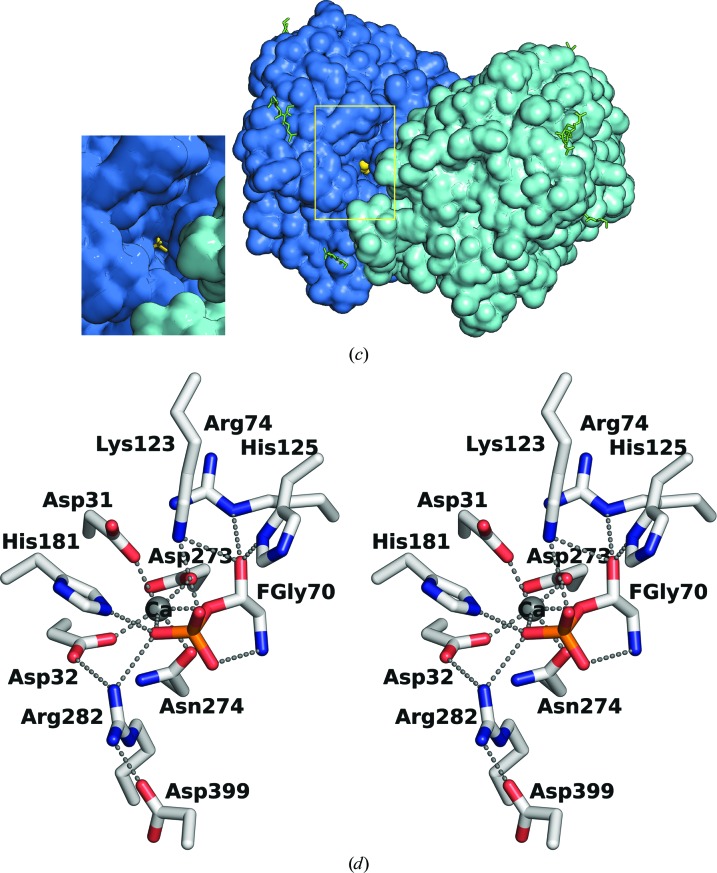
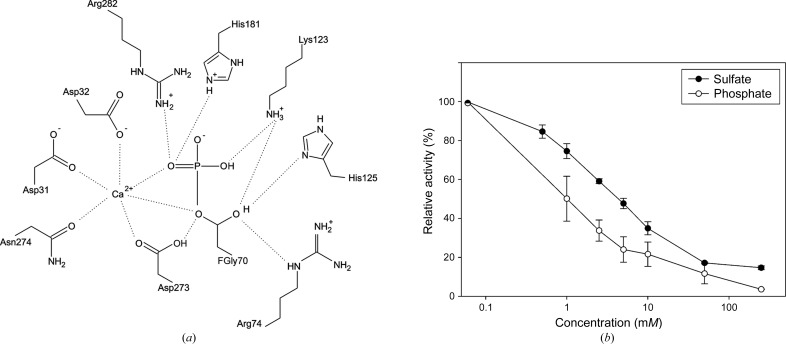
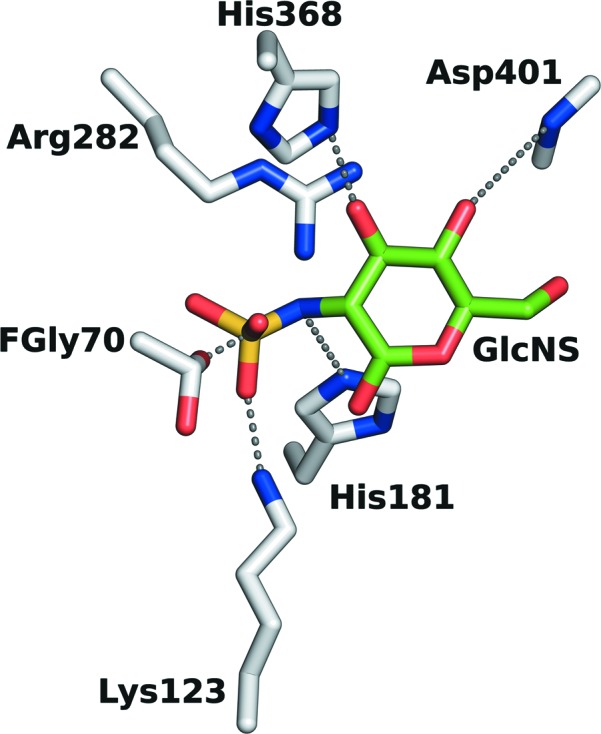
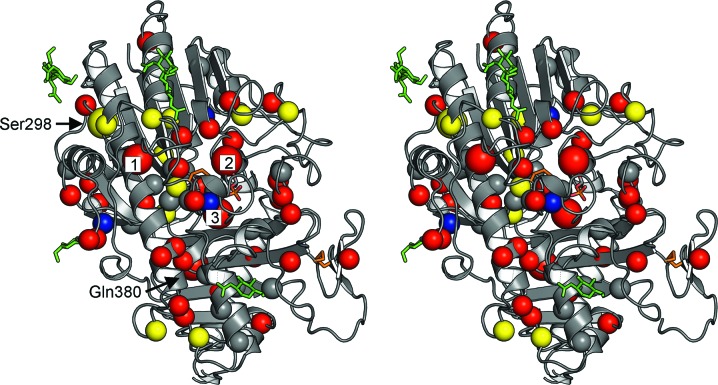
Mucopolysaccharidosis type IIIA (Sanfilippo A syndrome), a fatal childhood-onset neurodegenerative disease with mild facial, visceral and skeletal abnormalities, is caused by an inherited deficiency of the enzyme N-sulfoglucosamine sulfohydrolase (SGSH; sulfamidase). More than 100 mutations in the SGSH gene have been found to reduce or eliminate its enzymatic activity. However, the molecular understanding of the effect of these mutations has been confined by a lack of structural data for this enzyme. Here, the crystal structure of glycosylated SGSH is presented at 2 Å resolution. Despite the low sequence identity between this unique N-sulfatase and the group of O-sulfatases, they share a similar overall fold and active-site architecture, including a catalytic formylglycine, a divalent metal-binding site and a sulfate-binding site. However, a highly conserved lysine in O-sulfatases is replaced in SGSH by an arginine (Arg282) that is positioned to bind the N-linked sulfate substrate. The structure also provides insight into the diverse effects of pathogenic mutations on SGSH function in mucopolysaccharidosis type IIIA and convincing evidence for the molecular consequences of many missense mutations. Further, the molecular characterization of SGSH mutations will lay the groundwork for the development of structure-based drug design for this devastating neurodegenerative disorder.
Keywords: mucopolysaccharidosis IIIA; sulfamidase.
Figures
Similar articles
-
Reduction in Brain Heparan Sulfate with Systemic Administration of an IgG Trojan Horse-Sulfamidase Fusion Protein in the Mucopolysaccharidosis Type IIIA Mouse.Mol Pharm. 2018 Feb 5;15(2):602-608. doi: 10.1021/acs.molpharmaceut.7b00958. Epub 2017 Dec 29. Mol Pharm. 2018. PMID: 29251941
-
Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes.Hum Mol Genet. 2007 Nov 15;16(22):2693-702. doi: 10.1093/hmg/ddm223. Epub 2007 Aug 27. Hum Mol Genet. 2007. PMID: 17725987
-
Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells.Mol Pharm. 2014 Aug 4;11(8):2928-34. doi: 10.1021/mp500258p. Epub 2014 Jun 26. Mol Pharm. 2014. PMID: 24949884 Free PMC article.
-
Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications.Hum Mutat. 2001 Oct;18(4):264-81. doi: 10.1002/humu.1189. Hum Mutat. 2001. PMID: 11668611 Review.
-
Three-dimensional structures of sulfatases.Methods Enzymol. 2005;400:273-93. doi: 10.1016/S0076-6879(05)00016-9. Methods Enzymol. 2005. PMID: 16399355 Review.
Cited by
-
Bioinformatics classification of mutations in patients with Mucopolysaccharidosis IIIA.Metab Brain Dis. 2019 Dec;34(6):1577-1594. doi: 10.1007/s11011-019-00465-6. Epub 2019 Aug 5. Metab Brain Dis. 2019. PMID: 31385193 Free PMC article.
-
Impact of chemical modification of sulfamidase on distribution to brain interstitial fluid and to CSF after an intravenous administration in awake, freely-moving rats.Mol Genet Metab Rep. 2019 Dec 27;22:100554. doi: 10.1016/j.ymgmr.2019.100554. eCollection 2020 Mar. Mol Genet Metab Rep. 2019. PMID: 31908953 Free PMC article.
-
VarMeter2: An enhanced structure-based method for predicting pathogenic missense variants through Mahalanobis distance.Comput Struct Biotechnol J. 2025 Mar 1;27:1034-1047. doi: 10.1016/j.csbj.2025.02.008. eCollection 2025. Comput Struct Biotechnol J. 2025. PMID: 40160862 Free PMC article.
-
Sulfated glycan recognition by carbohydrate sulfatases of the human gut microbiota.Nat Chem Biol. 2022 Aug;18(8):841-849. doi: 10.1038/s41589-022-01039-x. Epub 2022 Jun 16. Nat Chem Biol. 2022. PMID: 35710619 Free PMC article.
-
Genetic Insights and Diagnostic Challenges in Highly Attenuated Lysosomal Storage Disorders.Genes (Basel). 2025 Jul 30;16(8):915. doi: 10.3390/genes16080915. Genes (Basel). 2025. PMID: 40869962 Free PMC article. Review.
References
-
- Baehner, F., Schmiedeskamp, C., Krummenauer, F., Miebach, E., Bajbouj, M., Whybra, C., Kohlschütter, A., Kampmann, C. & Beck, M. (2005). J. Inherit. Metab. Dis. 28, 1011–1017. - PubMed
-
- Ballabio, A. & Gieselmann, V. (2009). Biochim. Biophys. Acta, 1793, 684–696. - PubMed
-
- Bekri, S., Armana, G., De Ricaud, D., Osenda, M., Maire, I., Van Obberghen, E. & Froissart, R. (2005). J. Inherit. Metab. Dis. 28, 601–602. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
