

Arrhythmogenic cardiomyopathy: diagnosis, genetic background, and risk management
- PMID: 24817548
- PMCID: PMC4099433
- DOI: 10.1007/s12471-014-0563-7
Arrhythmogenic cardiomyopathy: diagnosis, genetic background, and risk management
Abstract
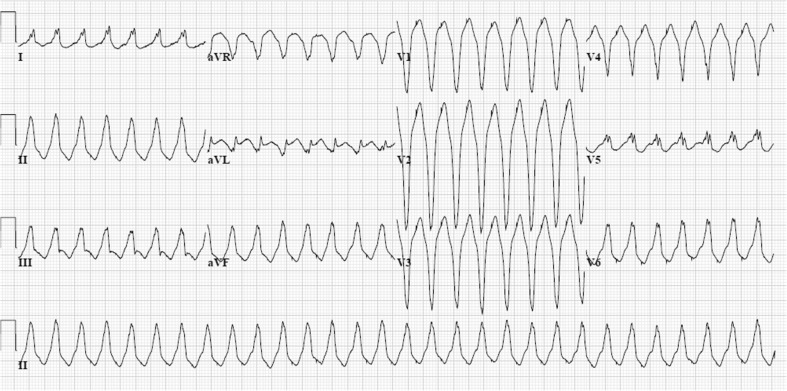
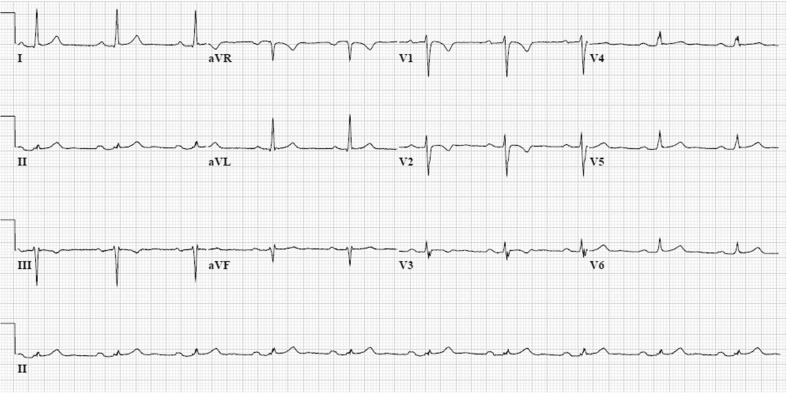
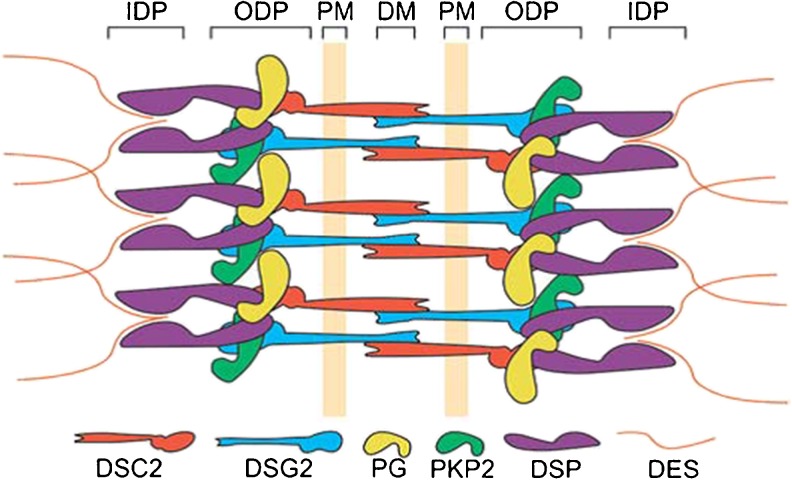
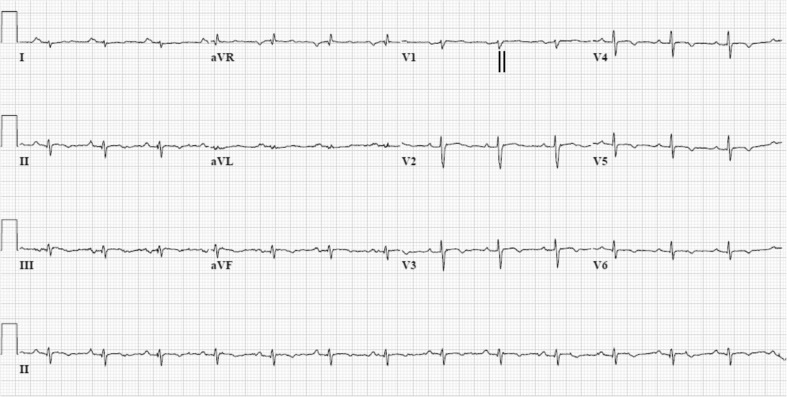
Arrhythmogenic cardiomyopathy (AC), also known as arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), is a hereditary disease characterised by ventricular arrhythmias, right ventricular and/or left ventricular dysfunction, and fibrofatty replacement of cardiomyocytes. Patients with AC typically present between the second and the fourth decade of life with ventricular tachycardias. However, sudden cardiac death (SCD) may be the first manifestation, often at young age in the concealed stage of disease. AC is diagnosed by a set of clinically applicable criteria defined by an international Task Force. The current Task Force Criteria are the essential standard for a correct diagnosis in individuals suspected of AC. The genetic substrate for AC is predominantly identified in genes encoding desmosomal proteins. In a minority of patients a non-desmosomal mutation predisposes to the phenotype. Risk stratification in AC is imperfect at present. Genotype-phenotype correlation analysis may provide more insight into risk profiles of index patients and family members. In addition to symptomatic treatment, prevention of SCD is the most important therapeutic goal in AC. Therapeutic options in symptomatic patients include antiarrhythmic drugs, catheter ablation, and ICD implantation. Furthermore, patients with AC and also all pathogenic mutation carriers should be advised against practising competitive and endurance sports.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
