

Overview of the genetic determinants of primary aldosteronism
- PMID: 24817817
- PMCID: PMC4012345
- DOI: 10.2147/TACG.S45620
Overview of the genetic determinants of primary aldosteronism
Abstract
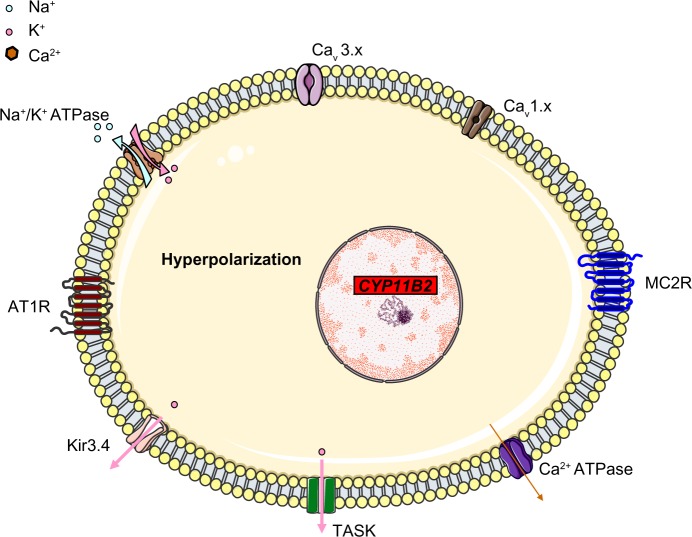
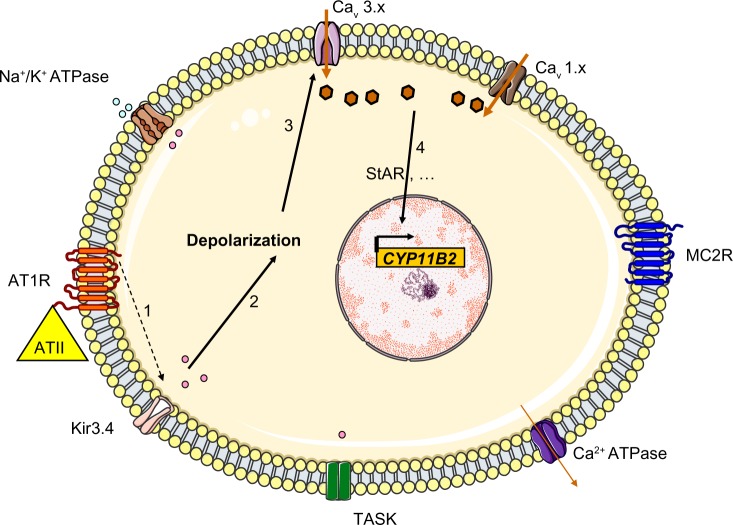
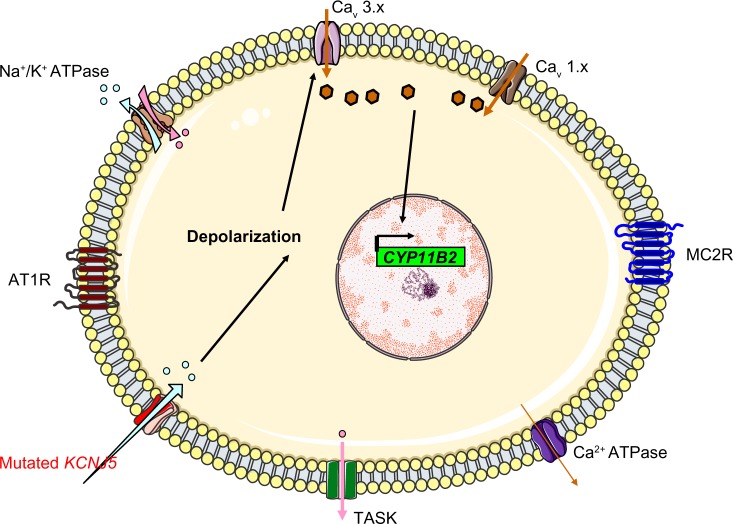
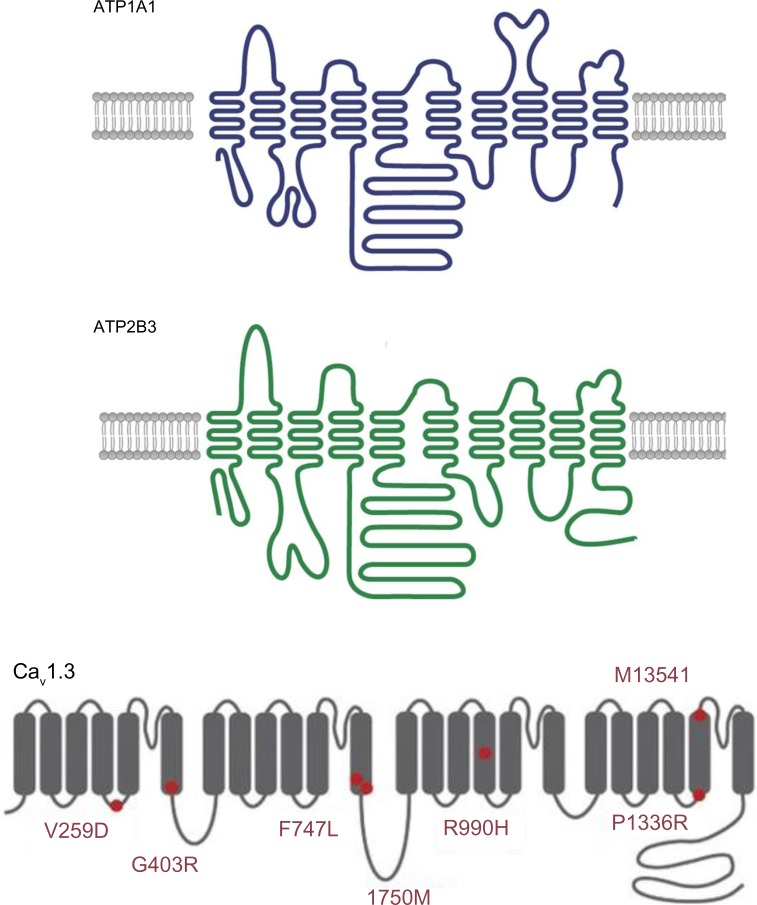
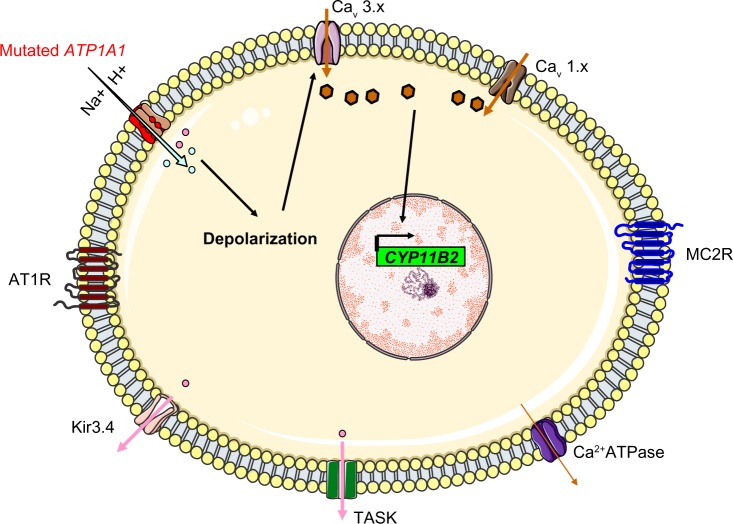
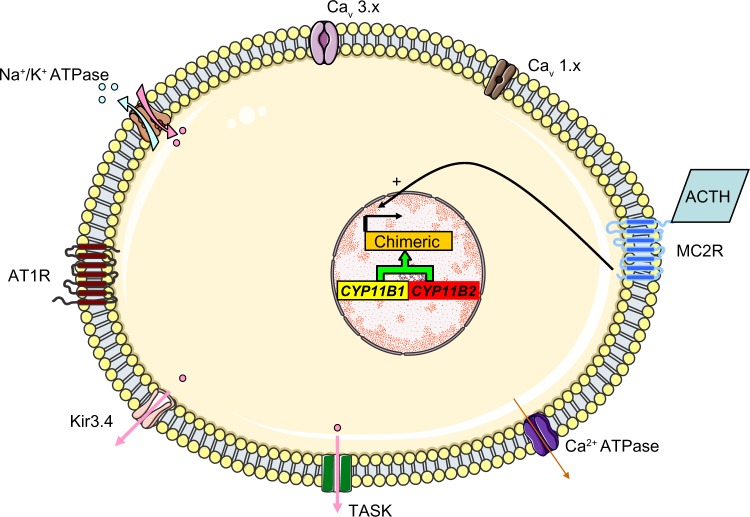
Primary aldosteronism is the most common cause of secondary hypertension. The syndrome accounts for 10% of all cases of hypertension and is primarily caused by bilateral adrenal hyperplasia or aldosterone-producing adenoma. Over the last few years, the use of exome sequencing has significantly improved our understanding of this syndrome. Somatic mutations in the KCNJ5, ATP1A1, ATP2B3 or CACNA1D genes are present in more than half of all cases of aldosterone-producing adenoma (~40%, ~6%, ~1% and ~8%, respectively). Germline gain-of-function mutations in KCNJ5 are now known to cause familial hyperaldosteronism type III, and an additional form of genetic hyperaldosteronism has been reported in patients with germline mutations in CACNA1D. These genes code for channels that control ion homeostasis across the plasma membrane of zona glomerulosa cells. Moreover, all these mutations modulate the same pathway, in which elevated intracellular calcium levels lead to aldosterone hyperproduction and (in some cases) adrenal cell proliferation. From a clinical standpoint, the discovery of these mutations has potential implications for patient management. The mutated channels could be targeted by drugs, in order to control hormonal and overgrowth-related manifestations. Furthermore, some of these mutations are associated with high cell turnover and may be amenable to diagnosis via the sequencing of cell-free (circulating) DNA. However, genotype-phenotype correlations in patients harboring these mutations have yet to be characterized. Despite this recent progress, much remains to be done to elucidate the yet unknown mechanisms underlying sporadic bilateral adrenal hyperplasia.
Keywords: potassium channels; primary aldosteronism; secondary hypertension.
Figures
References
-
- Funder JW, Carey RM, Fardella C, et al. Endocrine Society Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2008;93(9):3266–3281. - PubMed
-
- Rossi GP, Seccia TM, Pessina AC. Primary aldosteronism: part II: subtype differentiation and treatment. J Nephrol. 2008;21(4):455–462. - PubMed
-
- Funder JW. The genetics of primary aldosteronism: chapter two. Hypertension. 2012;59(3):537–538. - PubMed
-
- Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med. 1955;45(1):3–17. - PubMed
-
- Rossi GP, Bernini G, Caliumi C, et al. PAPY Study Investigators A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol. 2006;48(11):2293–2300. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
