

DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe
- PMID: 24819595
- PMCID: PMC4135522
- DOI: 10.1016/j.dnarep.2014.04.008
DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe
Abstract
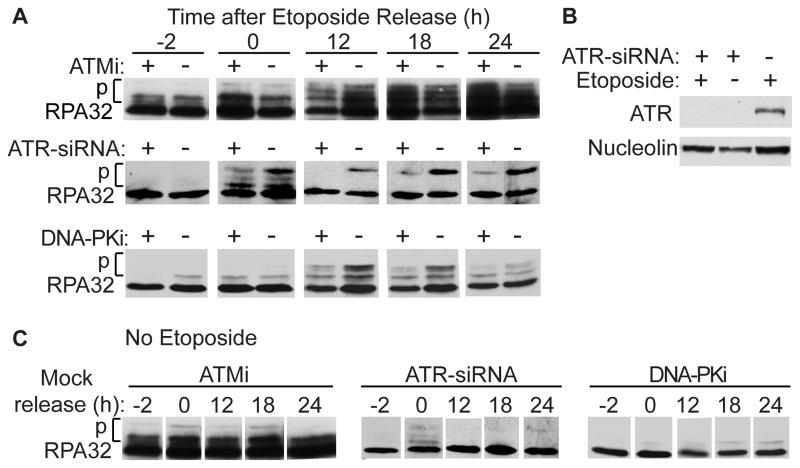
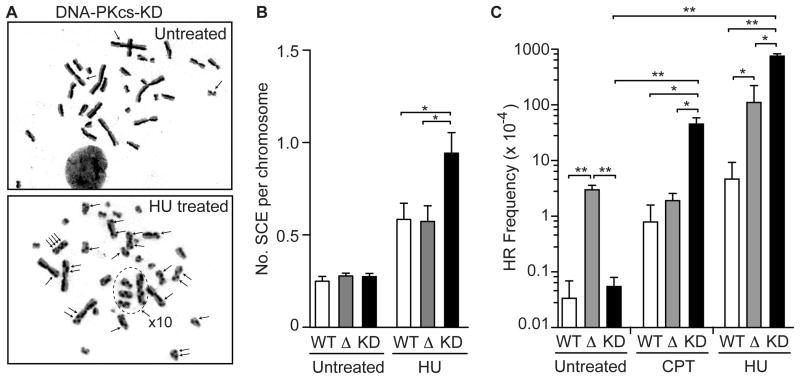
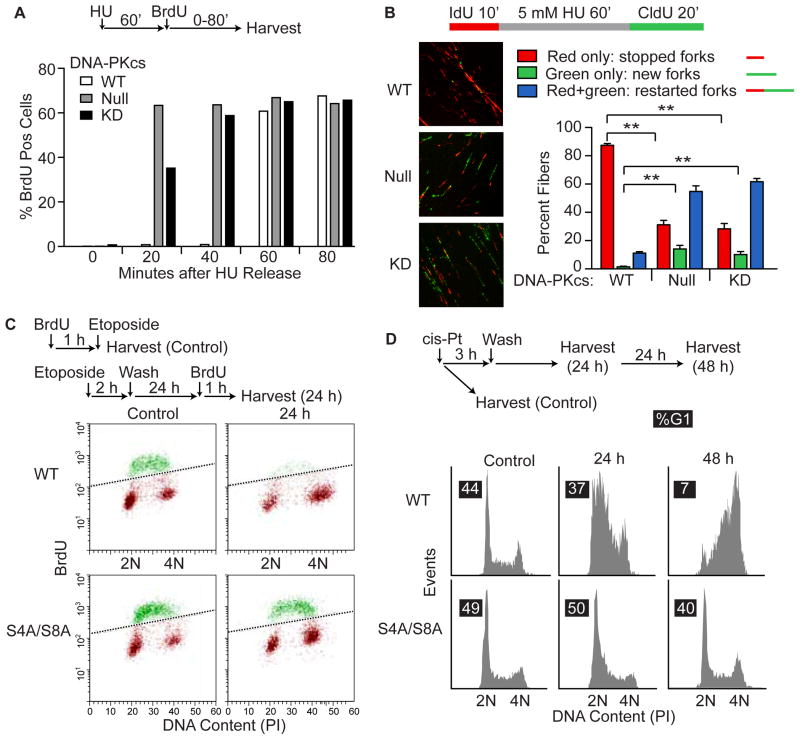
Genotoxins and other factors cause replication stress that activate the DNA damage response (DDR), comprising checkpoint and repair systems. The DDR suppresses cancer by promoting genome stability, and it regulates tumor resistance to chemo- and radiotherapy. Three members of the phosphatidylinositol 3-kinase-related kinase (PIKK) family, ATM, ATR, and DNA-PK, are important DDR proteins. A key PIKK target is replication protein A (RPA), which binds single-stranded DNA and functions in DNA replication, DNA repair, and checkpoint signaling. An early response to replication stress is ATR activation, which occurs when RPA accumulates on ssDNA. Activated ATR phosphorylates many targets, including the RPA32 subunit of RPA, leading to Chk1 activation and replication arrest. DNA-PK also phosphorylates RPA32 in response to replication stress, and we demonstrate that cells with DNA-PK defects, or lacking RPA32 Ser4/Ser8 targeted by DNA-PK, confer similar phenotypes, including defective replication checkpoint arrest, hyper-recombination, premature replication fork restart, failure to block late origin firing, and increased mitotic catastrophe. We present evidence that hyper-recombination in these mutants is ATM-dependent, but the other defects are ATM-independent. These results indicate that DNA-PK and ATR signaling through RPA32 plays a critical role in promoting genome stability and cell survival in response to replication stress.
Keywords: Checkpoint regulation; DNA repair; Homologous recombination; Replication stress.
Copyright © 2014 Elsevier B.V. All rights reserved.
Figures


Similar articles
-
Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress.Nucleic Acids Res. 2012 Nov;40(21):10780-94. doi: 10.1093/nar/gks849. Epub 2012 Sep 12. Nucleic Acids Res. 2012. PMID: 22977173 Free PMC article.
-
Role of RPA Phosphorylation in the ATR-Dependent G2 Cell Cycle Checkpoint.Genes (Basel). 2023 Dec 13;14(12):2205. doi: 10.3390/genes14122205. Genes (Basel). 2023. PMID: 38137027 Free PMC article.
-
DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair.Oncogene. 2013 May 9;32(19):2452-62. doi: 10.1038/onc.2012.257. Epub 2012 Jul 16. Oncogene. 2013. PMID: 22797063 Free PMC article.
-
Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway.Annu Rev Genet. 2016 Nov 23;50:155-173. doi: 10.1146/annurev-genet-121415-121658. Epub 2016 Sep 9. Annu Rev Genet. 2016. PMID: 27617969 Review.
-
Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition.Cancer Treat Rev. 2018 Dec;71:1-7. doi: 10.1016/j.ctrv.2018.09.003. Epub 2018 Sep 11. Cancer Treat Rev. 2018. PMID: 30269007 Free PMC article. Review.
Cited by
-
DDX41 coordinates RNA splicing and transcriptional elongation to prevent DNA replication stress in hematopoietic cells.Leukemia. 2022 Nov;36(11):2605-2620. doi: 10.1038/s41375-022-01708-9. Epub 2022 Oct 14. Leukemia. 2022. PMID: 36229594 Free PMC article.
-
DNA-PK, ATM, and ATR: PIKKing on p53.Cell Cycle. 2018;17(3):275-276. doi: 10.1080/15384101.2017.1412147. Epub 2018 Apr 2. Cell Cycle. 2018. PMID: 29265907 Free PMC article. No abstract available.
-
Modulation of DNA damage and repair pathways by human tumour viruses.Viruses. 2015 May 22;7(5):2542-91. doi: 10.3390/v7052542. Viruses. 2015. PMID: 26008701 Free PMC article. Review.
-
G9a coordinates with the RPA complex to promote DNA damage repair and cell survival.Proc Natl Acad Sci U S A. 2017 Jul 25;114(30):E6054-E6063. doi: 10.1073/pnas.1700694114. Epub 2017 Jul 11. Proc Natl Acad Sci U S A. 2017. PMID: 28698370 Free PMC article.
-
An ATM-PPM1D Circuit Controls the Processing and Restart of DNA Replication Forks.bioRxiv [Preprint]. 2025 May 15:2025.05.13.652823. doi: 10.1101/2025.05.13.652823. bioRxiv. 2025. PMID: 40462982 Free PMC article. Preprint.
References
-
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, Casellas R, Robbiani DF, Staudt L, Fernandez-Capetillo O, Nussenzweig A. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. - PMC - PubMed
-
- Shen Z, Nickoloff JA. Mammalian homologous recombination repair and cancer intervention. In: Wei Q, Li L, Chen DJ, editors. DNA Repair, Genetic Instability, and Cancer. World Scientific Publishing Co; Singapore: 2007. pp. 119–156.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
