

High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution
- PMID: 24820965
- PMCID: PMC4018626
- DOI: 10.1038/srep04942
High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution
Abstract
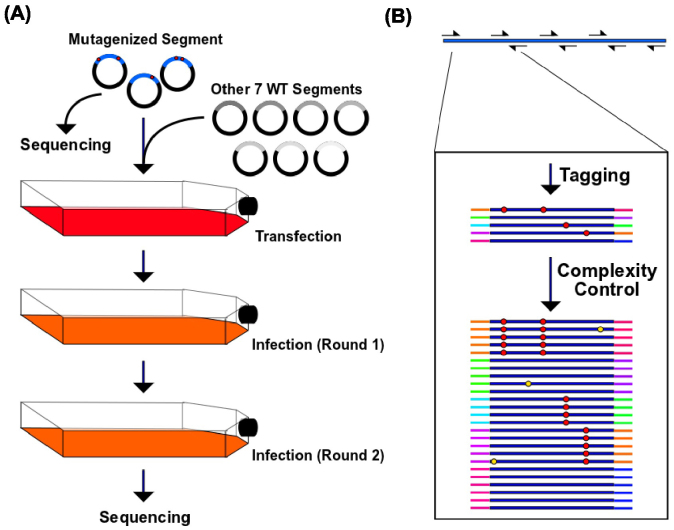
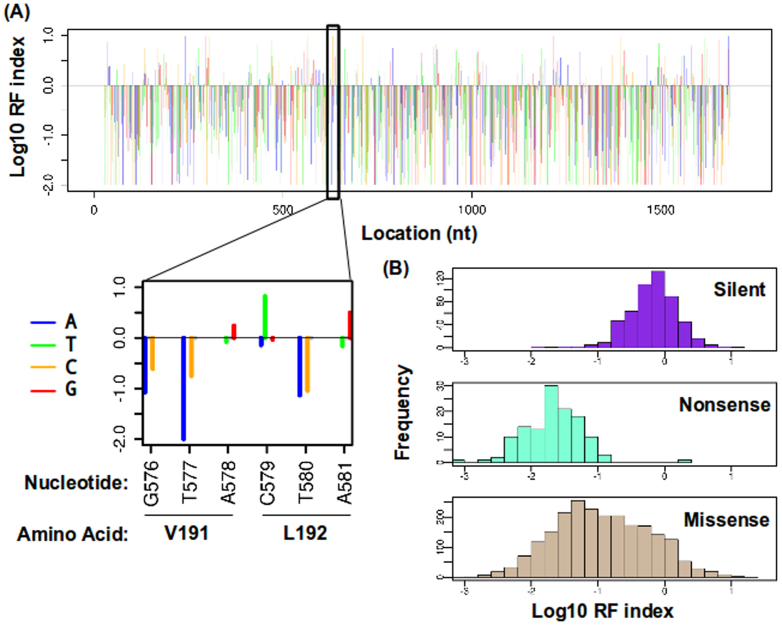
Genetic research on influenza virus biology has been informed in large part by nucleotide variants present in seasonal or pandemic samples, or individual mutants generated in the laboratory, leaving a substantial part of the genome uncharacterized. Here, we have developed a single-nucleotide resolution genetic approach to interrogate the fitness effect of point mutations in 98% of the amino acid positions in the influenza A virus hemagglutinin (HA) gene. Our HA fitness map provides a reference to identify indispensable regions to aid in drug and vaccine design as targeting these regions will increase the genetic barrier for the emergence of escape mutations. This study offers a new platform for studying genome dynamics, structure-function relationships, virus-host interactions, and can further rational drug and vaccine design. Our approach can also be applied to any virus that can be genetically manipulated.
Figures
References
-
- Mardis E. R. Next-generation dna sequencing methods. Annu Rev Genomics Hum Genet 9, 387–402 (2008). - PubMed
-
- Schena M., Shalon D., Davis R. W. & Brown P. O. Quantitative monitoring of gene expression patterns with a complementary dna microarray. Science 270, 467–470 (1995). - PubMed
-
- Barski A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
