

Genome scale modeling in systems biology: algorithms and resources
- PMID: 24822031
- PMCID: PMC4009841
- DOI: 10.2174/1389202915666140319002221
Genome scale modeling in systems biology: algorithms and resources
Abstract
In recent years, in silico studies and trial simulations have complemented experimental procedures. A model is a description of a system, and a system is any collection of interrelated objects; an object, moreover, is some elemental unit upon which observations can be made but whose internal structure either does not exist or is ignored. Therefore, any network analysis approach is critical for successful quantitative modeling of biological systems. This review highlights some of most popular and important modeling algorithms, tools, and emerging standards for representing, simulating and analyzing cellular networks in five sections. Also, we try to show these concepts by means of simple example and proper images and graphs. Overall, systems biology aims for a holistic description and understanding of biological processes by an integration of analytical experimental approaches along with synthetic computational models. In fact, biological networks have been developed as a platform for integrating information from high to low-throughput experiments for the analysis of biological systems. We provide an overview of all processes used in modeling and simulating biological networks in such a way that they can become easily understandable for researchers with both biological and mathematical backgrounds. Consequently, given the complexity of generated experimental data and cellular networks, it is no surprise that researchers have turned to computer simulation and the development of more theory-based approaches to augment and assist in the development of a fully quantitative understanding of cellular dynamics.
Keywords: Biological network.; Genome-scale modeling; Modeling algorithms; Systems biology.
Figures

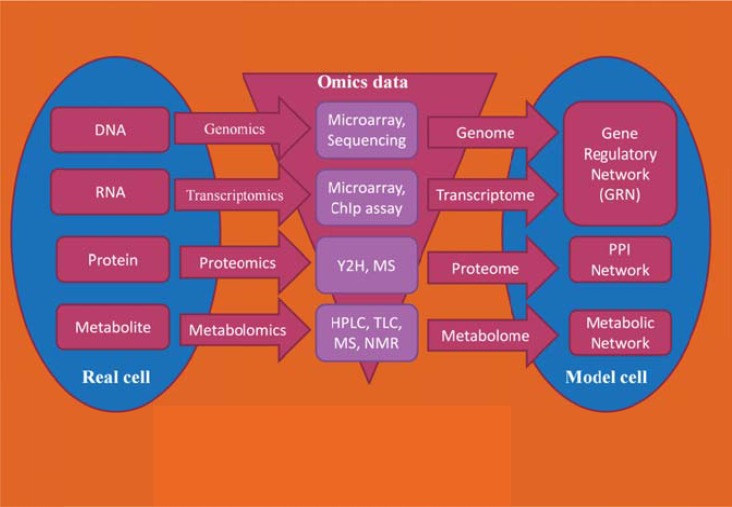
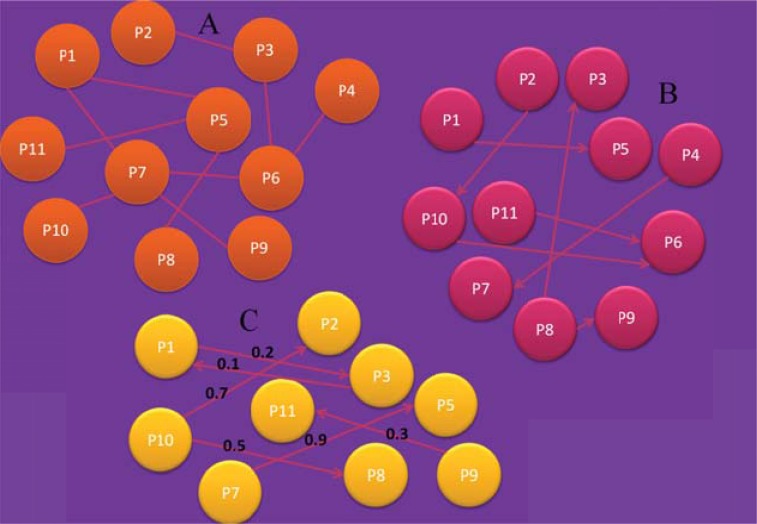
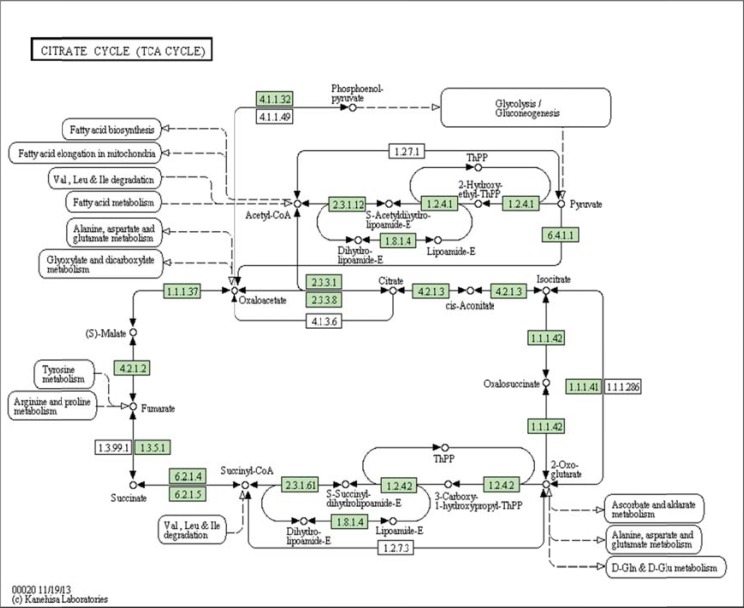
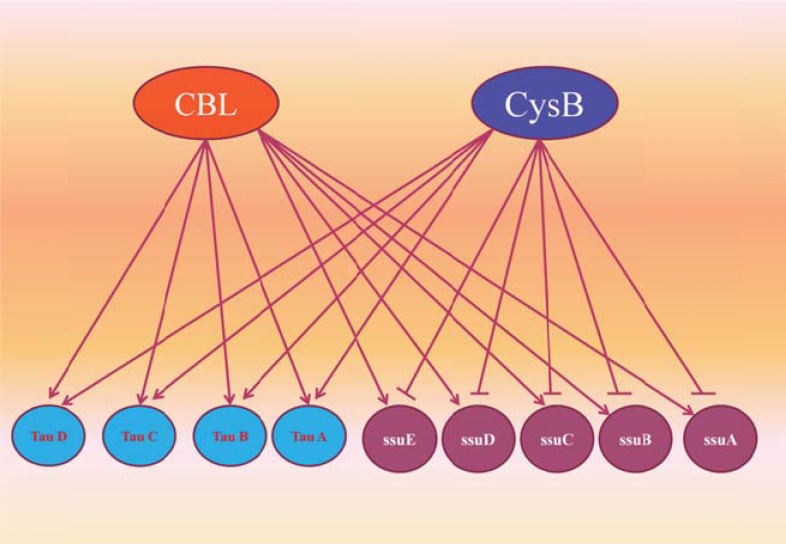
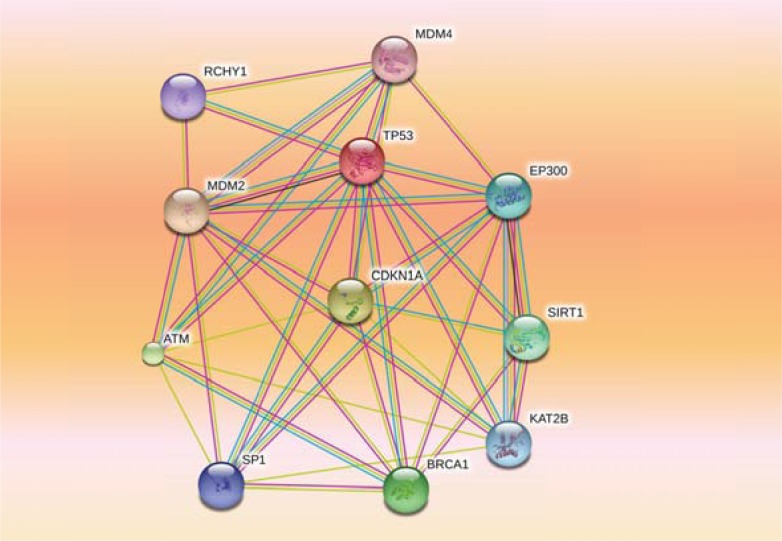

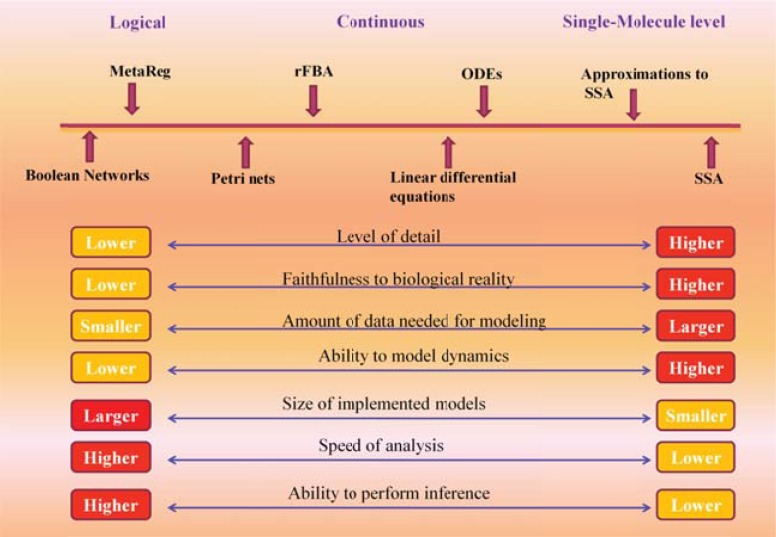
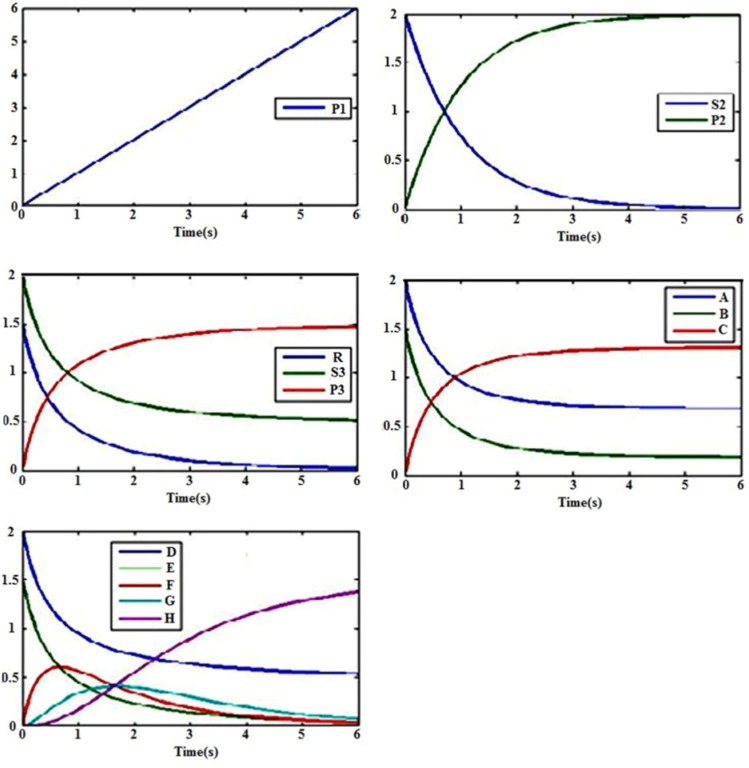
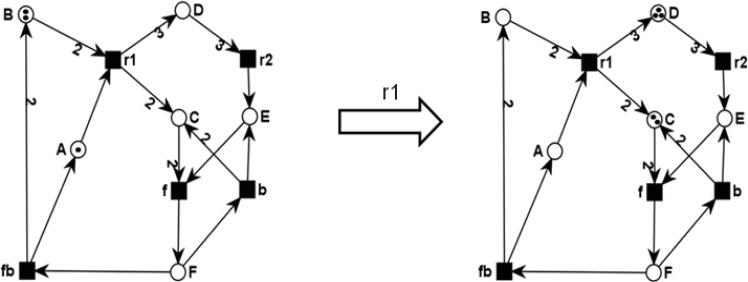

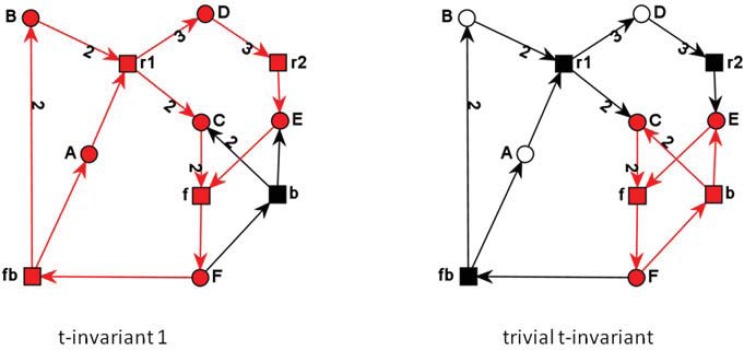

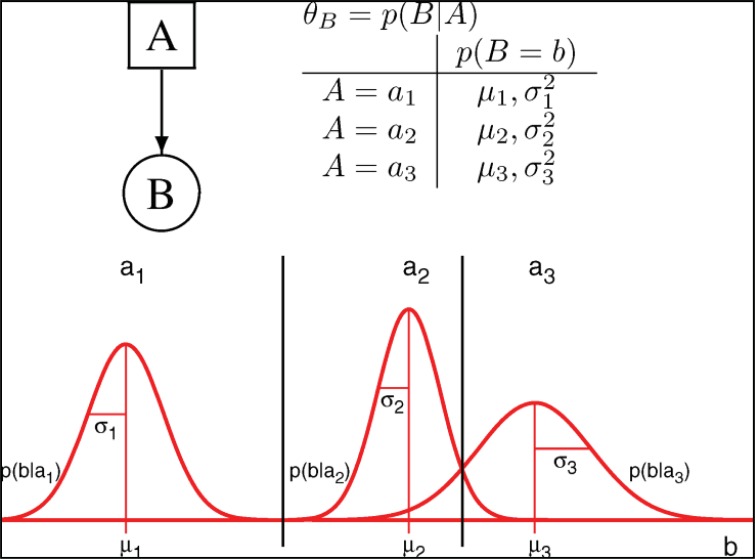
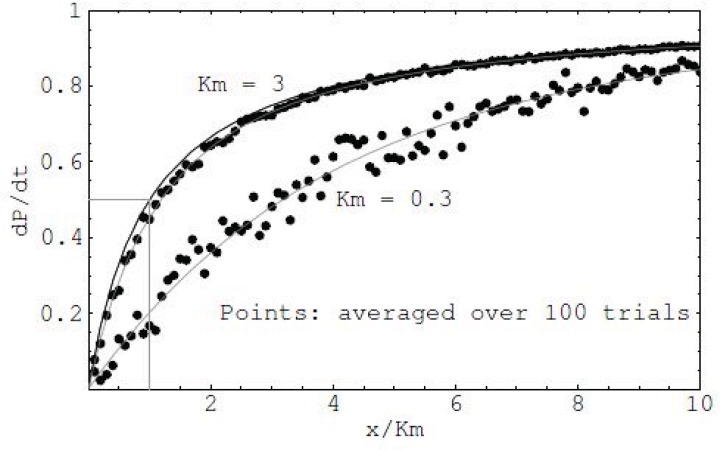
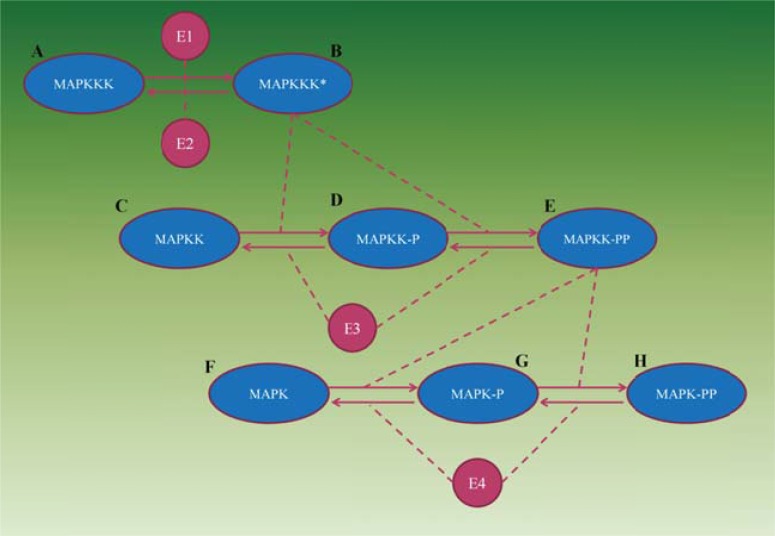

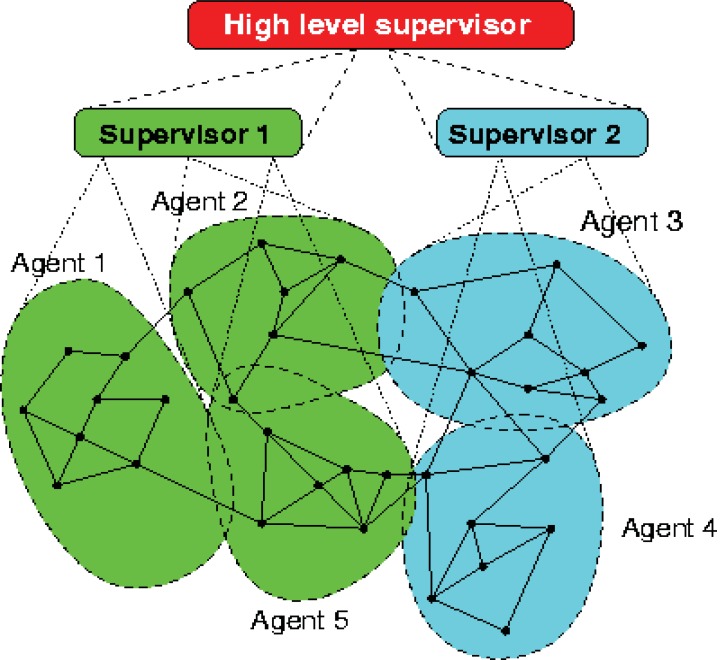


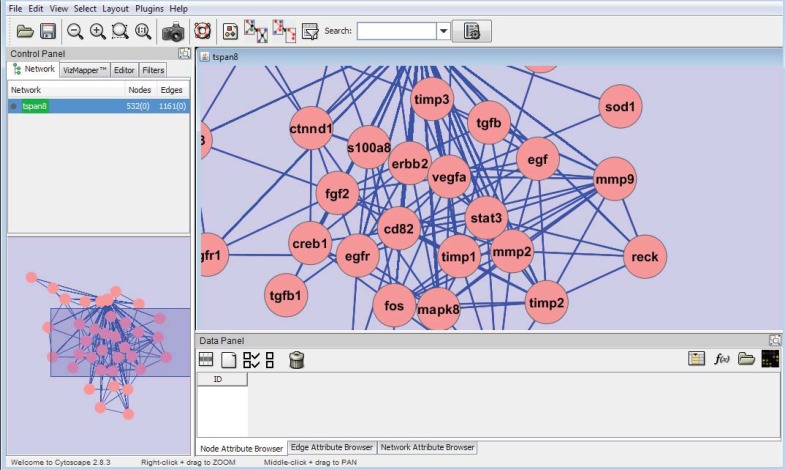
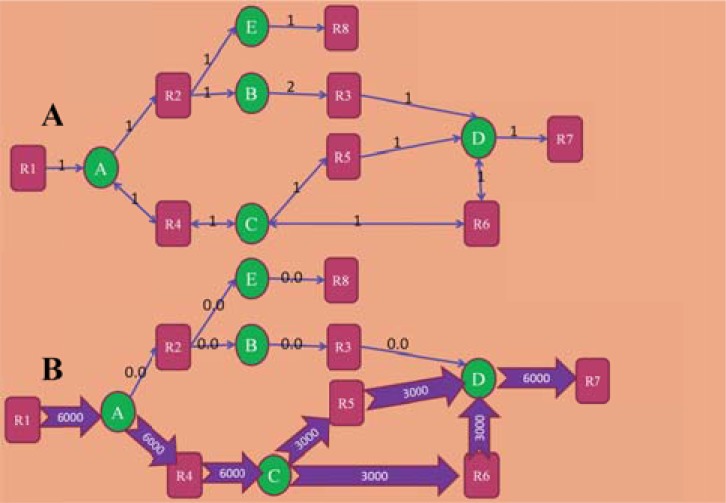

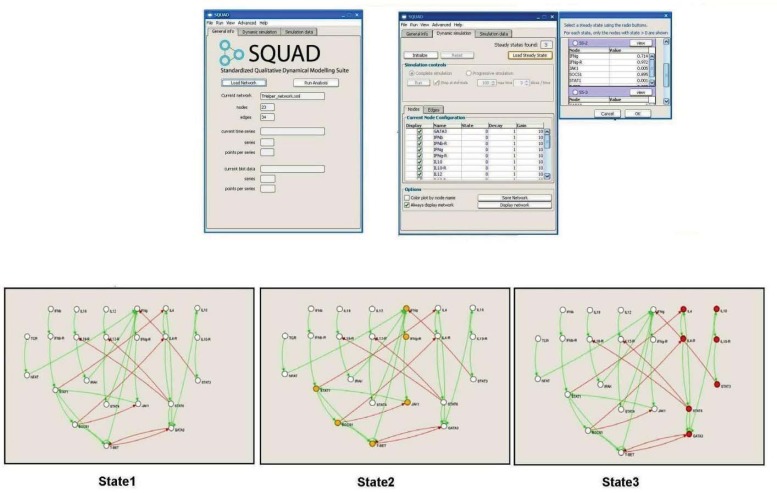

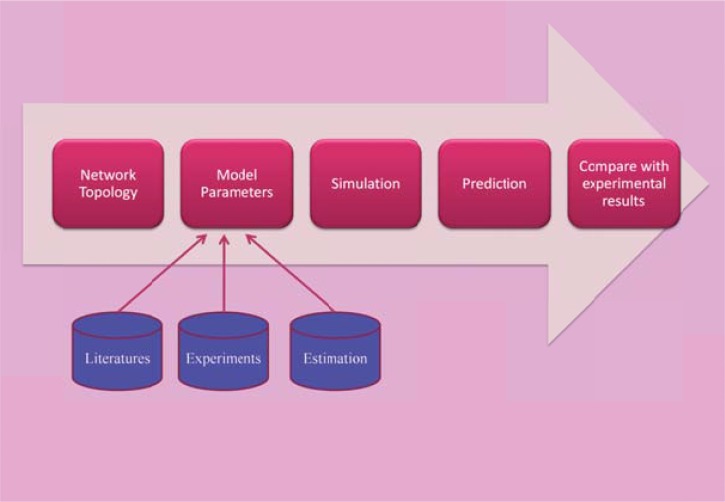
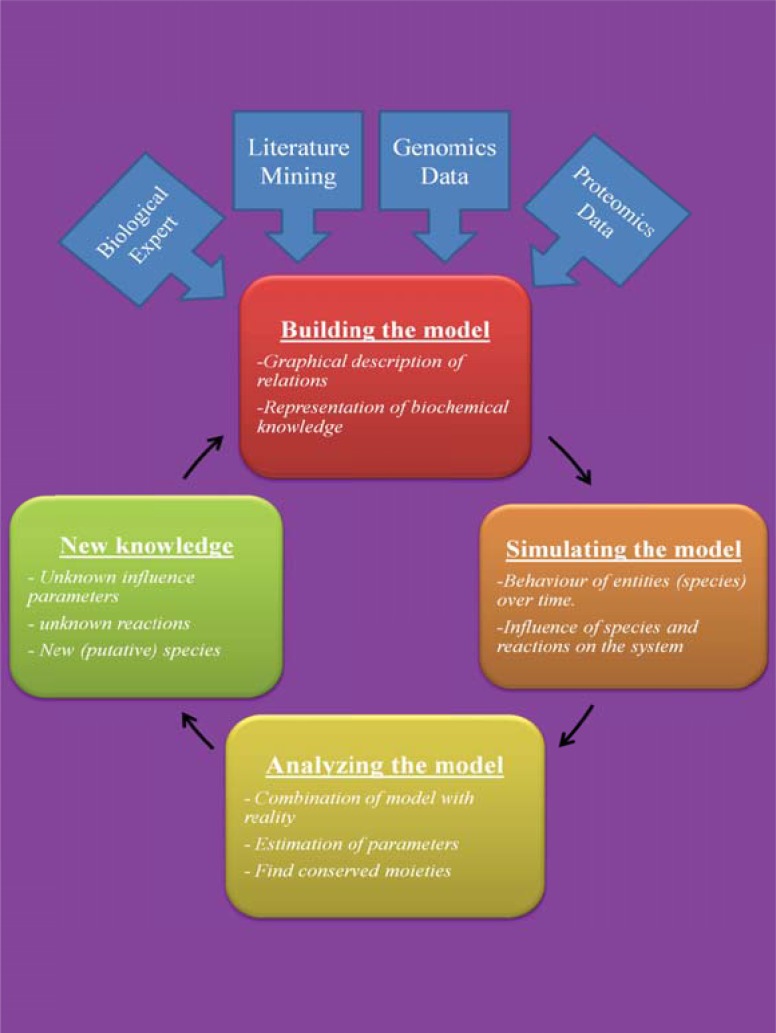
References
-
- Kitano H. Systems biology: a brief overview. Science. 2002;295(5560):1662–4. - PubMed
-
- Friboulet A, Thomas D. Systems biology—an interdisciplinary approach. Biosens. Bioelectron. 2005;20(12):2404–2407. - PubMed
-
- Laval J M, Mazeran P E, Thomas D. Nanobiotechnology and its role in the development of new analytical devices. Analyst. 2000;125(1):29–33. - PubMed
-
- Westerhoff H V, Palsson B O. The evolution of molecular biology into systems biology. Nat. Biotechnol. 2004;22(10):1249–52. - PubMed
-
- (a) Covert M W, Schilling C H, Famili I, Edwards J S, Goryanin II, Selkov E, Palsson B O. Metabolic modeling of microbial strains in silico. Trends Biochem. Sci. 2001;26(3):179–86. - PubMed
- (b) Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat. Chem. Biol. 2010;6(11):787–9. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources