

A rotamer library to enable modeling and design of peptoid foldamers
- PMID: 24823488
- PMCID: PMC4227732
- DOI: 10.1021/ja503776z
A rotamer library to enable modeling and design of peptoid foldamers
Abstract
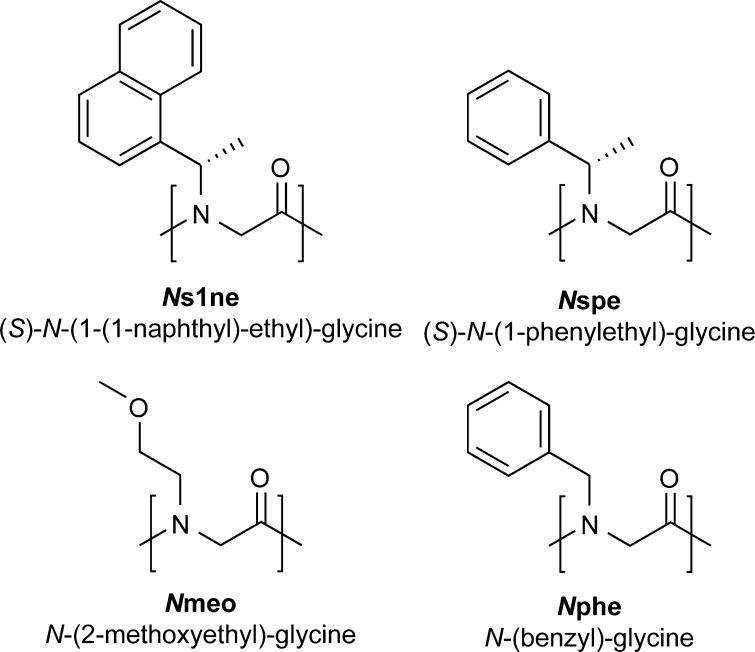
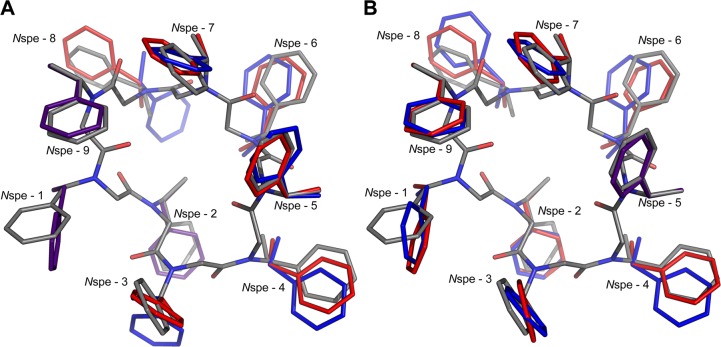
Peptoids are a family of synthetic oligomers composed of N-substituted glycine units. Along with other "foldamer" systems, peptoid oligomer sequences can be predictably designed to form a variety of stable secondary structures. It is not yet evident if foldamer design can be extended to reliably create tertiary structure features that mimic more complex biomolecular folds and functions. Computational modeling and prediction of peptoid conformations will likely play a critical role in enabling complex biomimetic designs. We introduce a computational approach to provide accurate conformational and energetic parameters for peptoid side chains needed for successful modeling and design. We find that peptoids can be described by a "rotamer" treatment, similar to that established for proteins, in which the peptoid side chains display rotational isomerism to populate discrete regions of the conformational landscape. Because of the insufficient number of solved peptoid structures, we have calculated the relative energies of side-chain conformational states to provide a backbone-dependent (BBD) rotamer library for a set of 54 different peptoid side chains. We evaluated two rotamer library development methods that employ quantum mechanics (QM) and/or molecular mechanics (MM) energy calculations to identify side-chain rotamers. We show by comparison to experimental peptoid structures that both methods provide an accurate prediction of peptoid side chain placements in folded peptoid oligomers and at protein interfaces. We have incorporated our peptoid rotamer libraries into ROSETTA, a molecular design package previously validated in the context of protein design and structure prediction.
Figures







Similar articles
-
Construction of peptoids with all trans-amide backbones and peptoid reverse turns via the tactical incorporation of N-aryl side chains capable of hydrogen bonding.J Org Chem. 2010 Sep 17;75(18):6068-78. doi: 10.1021/jo101075a. J Org Chem. 2010. PMID: 20722367 Free PMC article.
-
β-Peptoid Foldamers at Last.Acc Chem Res. 2015 Oct 20;48(10):2696-704. doi: 10.1021/acs.accounts.5b00257. Epub 2015 Jul 15. Acc Chem Res. 2015. PMID: 26176689
-
New strategies for the design of folded peptoids revealed by a survey of noncovalent interactions in model systems.J Am Chem Soc. 2009 Nov 18;131(45):16555-67. doi: 10.1021/ja907184g. J Am Chem Soc. 2009. PMID: 19860427 Free PMC article.
-
Strategies to Control the Cis-Trans Isomerization of Peptoid Amide Bonds.Chem Asian J. 2022 Jun 1;17(11):e202200149. doi: 10.1002/asia.202200149. Epub 2022 Apr 20. Chem Asian J. 2022. PMID: 35362652 Review.
-
Helices in peptoids of alpha- and beta-peptides.Phys Biol. 2006 Feb 2;3(1):S1-9. doi: 10.1088/1478-3975/3/1/S01. Phys Biol. 2006. PMID: 16582460 Review.
Cited by
-
Protocols for Molecular Modeling with Rosetta3 and RosettaScripts.Biochemistry. 2016 Aug 30;55(34):4748-63. doi: 10.1021/acs.biochem.6b00444. Epub 2016 Aug 16. Biochemistry. 2016. PMID: 27490953 Free PMC article. Review.
-
Rotamer Libraries for the High-Resolution Design of β-Amino Acid Foldamers.Structure. 2017 Nov 7;25(11):1771-1780.e3. doi: 10.1016/j.str.2017.09.005. Epub 2017 Oct 12. Structure. 2017. PMID: 29033287 Free PMC article.
-
Artificial Intelligence Transforming Post-Translational Modification Research.Bioengineering (Basel). 2024 Dec 31;12(1):26. doi: 10.3390/bioengineering12010026. Bioengineering (Basel). 2024. PMID: 39851300 Free PMC article. Review.
-
A backbone-dependent rotamer library with high (ϕ, ψ) coverage using metadynamics simulations.Protein Sci. 2022 Dec;31(12):e4491. doi: 10.1002/pro.4491. Protein Sci. 2022. PMID: 36327064 Free PMC article.
-
Design of Peptoid-peptide Macrocycles to Inhibit the β-catenin TCF Interaction in Prostate Cancer.Nat Commun. 2018 Oct 23;9(1):4396. doi: 10.1038/s41467-018-06845-3. Nat Commun. 2018. PMID: 30352998 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
