

Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia
- PMID: 24825865
- PMCID: PMC4148768
- DOI: 10.1182/blood-2014-03-559542
Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia
Abstract
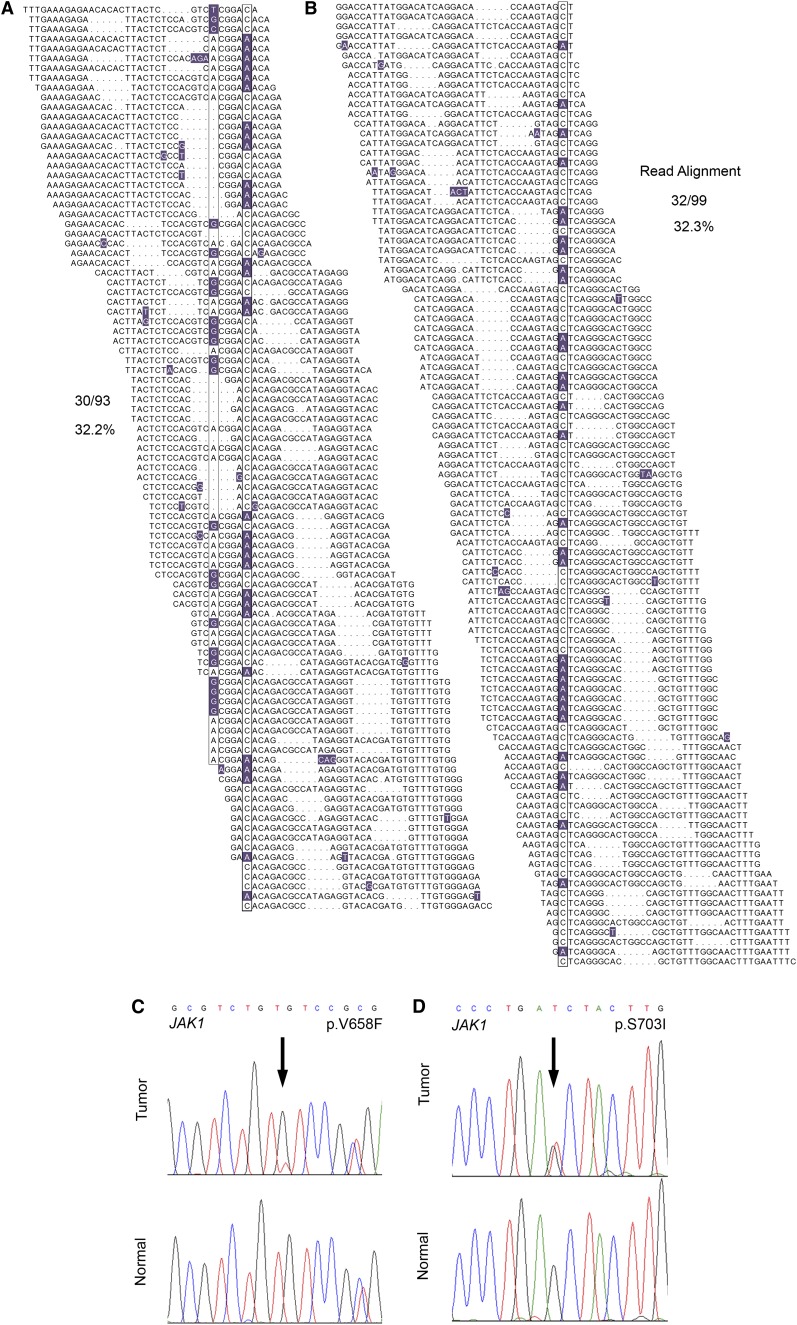
The comprehensive genetic alterations underlying the pathogenesis of T-cell prolymphocytic leukemia (T-PLL) are unknown. To address this, we performed whole-genome sequencing (WGS), whole-exome sequencing (WES), high-resolution copy-number analysis, and Sanger resequencing of a large cohort of T-PLL. WGS and WES identified novel mutations in recurrently altered genes not previously implicated in T-PLL including EZH2, FBXW10, and CHEK2. Strikingly, WGS and/or WES showed largely mutually exclusive mutations affecting IL2RG, JAK1, JAK3, or STAT5B in 38 of 50 T-PLL genomes (76.0%). Notably, gain-of-function IL2RG mutations are novel and have not been reported in any form of cancer. Further, high-frequency mutations in STAT5B have not been previously reported in T-PLL. Functionally, IL2RG-JAK1-JAK3-STAT5B mutations led to signal transducer and activator of transcription 5 (STAT5) hyperactivation, transformed Ba/F3 cells resulting in cytokine-independent growth, and/or enhanced colony formation in Jurkat T cells. Importantly, primary T-PLL cells exhibited constitutive activation of STAT5, and targeted pharmacologic inhibition of STAT5 with pimozide induced apoptosis in primary T-PLL cells. These results for the first time provide a portrait of the mutational landscape of T-PLL and implicate deregulation of DNA repair and epigenetic modulators as well as high-frequency mutational activation of the IL2RG-JAK1-JAK3-STAT5B axis in the pathogenesis of T-PLL. These findings offer opportunities for novel targeted therapies in this aggressive leukemia.
© 2014 by The American Society of Hematology.
Figures




Comment in
-
Take (STAT)5: jazzing up T-cell leukemia.Blood. 2014 Aug 28;124(9):1383-4. doi: 10.1182/blood-2014-06-576942. Blood. 2014. PMID: 25170111 Free PMC article.
References
-
- Matutes E, Brito-Babapulle V, Swansbury J, et al. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood. 1991;78(12):3269–3274. - PubMed
-
- Hopfinger G, Busch R, Pflug N, et al. Sequential chemoimmunotherapy of fludarabine, mitoxantrone, and cyclophosphamide induction followed by alemtuzumab consolidation is effective in T-cell prolymphocytic leukemia. Cancer. 2013;119(12):2258–2267. - PubMed
-
- Dearden CE. T-cell prolymphocytic leukemia. Clin Lymphoma Myeloma. 2009;(9)Suppl 3:S239-S243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
